基于R软件的Lasso回归在肿瘤信息基因选择中的应用
- 格式:pdf
- 大小:275.01 KB
- 文档页数:6

rlasso回归的原理Rlasso回归是一种统计方法,用于分析因变量(响应变量)和自变量(预测变量)之间的关系。
它是一种广义线性模型(GLM)的扩展,适用于连续或分类因变量,并具有很强的回归性能。
在本篇文章中,我们将介绍Rlasso回归的原理,包括其基本概念、应用场景、优缺点以及算法实现。
一、基本概念Rlasso回归将自变量和因变量视为两个随机向量,并使用广义线性模型(GLM)来拟合它们之间的关系。
在Rlasso回归中,因变量和自变量的关系可以表示为y~x的形式,其中y是连续因变量,x是自变量向量。
广义线性模型假设因变量的自然对数服从某个概率分布,如正态分布、泊松分布等。
通过拟合广义线性模型,Rlasso回归可以估计自变量对因变量的影响程度。
二、应用场景Rlasso回归适用于各种应用场景,包括但不限于以下几种:1.生物医学研究:在医学研究中,基因表达数据通常包含多个基因,可以通过Rlasso回归分析它们与疾病表型之间的关系。
2.市场研究:在市场研究中,可以通过Rlasso回归分析产品特性、价格、促销等因素与销售量之间的关系。
3.金融学研究:在金融学研究中,可以使用Rlasso回归分析股票价格、收益率、交易量等指标之间的关系。
三、优缺点1.适用于连续和分类因变量:Rlasso回归适用于因变量为连续或分类的情况,具有广泛的适用性。
2.强大的回归性能:与其他回归方法相比,Rlasso回归具有更好的回归性能,能够更好地拟合数据。
3.可解释性强:Rlasso回归可以通过模型参数来解释自变量对因变量的影响程度,便于理解和解释结果。
然而,Rlasso回归也存在一些缺点:1.需要较长的计算时间:与其他线性回归方法相比,Rlasso回归的计算时间较长。
2.受到正则化参数的影响:选择合适的正则化参数对于模型的拟合至关重要,参数选择不当可能会导致过拟合或欠拟合。
3.仅适用于连续自变量:Rlasso回归仅适用于自变量为连续的情况,对于分类自变量可能需要其他方法进行处理。

Lasso方法简要介绍及其在回归分析中的应用回归分析(Regression Analysis)是确定两种或两种以上变量间相互依赖的定量关系的一种统计分析方法。
最早形式的回归分析可以追溯到两百多年前由德国数学家高斯提出的最小二乘法。
而回归分析也是研究时间最长和应用最广泛的的方法。
自从产生以来回归分析一直都是统计学家研究的一个重点领域,直到近二十多年来还有很多对回归分析提出的各种新的改进。
回归模型一般假设响应变量(response variable)也叫自变量和独立变量(independent variables)也叫因变量,有具体的参数化(parametric)形式的关系,而这些参数有很多成熟的方法可以去估计(比如最小二乘法),误差分析方法也有详细的研究。
总的来说,回归分析方法具有数据适应性强,模型估计稳定,误差容易分析等优良特点,即使在机器学习方法发展如此多种多样的今天,依然是各个领域中最常用的分析方法之一。
回归分析中最常见的线性回归假设响应和独立变量间存在明显的线性关系。
如图一所示,响应变量(黑点)的数值大致在一条直线周围,除了每个点都有的随机误差。
线性回归模型看似极大的简化了响应变量和独立变量之间的关系,其实在实际分析中往往是最稳定的模型。
因为线性模型受到极端或者坏数据的影响最小。
例如预测病人的住院成本,很可能出现其中一两个病人会有很大的花费,这个可能是跟病理无关的,这种病人的数据就很可能影响整个模型对于一般病人住院成本的预测。
所以一个统计模型的稳定性是实际应用中的关键:对于相似的数据应该得出相似的分析结果。
这种稳定性一般统计里用模型的方差来表示,稳定性越好,模型的方差越小。
图1. 线性回归示意图在统计学习中存在一个重要理论:方差权衡。
一般常理认为模型建立得越复杂,分析和预测效果应该越好。
而方差权衡恰恰指出了其中的弊端。
复杂的模型一般对已知数据(training sample)的拟合(fitting)大过于简单模型,但是复杂模型很容易对数据出现过度拟合(over-fitting)。

lasso预测模型中四种用法
lasso 预测模型是一种常用的机器学习算法,有以下四种用法:
1. 筛选关键变量:利用 Lasso 回归筛选出与目标变量密切相关的病理生理学指标,相比于既往常用的逐步向前或向后回归,Lasso 回归具有更强的过滤能力,能更好地识别影响目标变量的关键变量。
2. 构建预测模型:根据随访数据构建新的预测模型,以识别高危人群,制定个性化的管理策略。
3. 减少变量数量:与全球急性冠状动脉事件注册(GRACE)评分相比,Lasso 预测模型能在纳入变量更少的情况下,提高预测效能,更适合在中国临床诊疗中推广。
4. 提高模型准确性:通过 Lasso 回归对数据进行降维,减少特征数量,提高模型的准确性,从而提高预测效果。
在实际应用中,可以根据具体需求选择合适的 Lasso 预测模型用法。

r语言lasso回归应用实例以R语言lasso回归应用实例为标题的文章引言在现代统计学中,回归分析是一种常用的方法,用于研究自变量与因变量之间的关系。
然而,传统的回归分析方法在处理高维数据时面临一些挑战,例如变量选择和模型解释的困难。
为了解决这些问题,lasso回归成为了一种流行的方法。
本文将介绍使用R语言进行lasso回归分析的应用实例。
数据准备我们需要准备一个数据集,以便进行lasso回归分析。
我们使用R 语言内置的mtcars数据集作为示例。
该数据集包含了32辆汽车的11个变量,包括汽车的性能指标和特征。
数据预处理在进行回归分析之前,我们需要对数据进行一些预处理操作。
首先,我们将数据集分为自变量和因变量。
在这个例子中,我们将mpg (每加仑行驶的英里数)作为因变量,将其他变量作为自变量。
```# 导入数据集data(mtcars)# 将mpg作为因变量,其他变量作为自变量X <- as.matrix(mtcars[, -1])Y <- mtcars[, 1]```Lasso回归分析接下来,我们使用R语言中的glmnet包进行lasso回归分析。
glmnet包是一个用于普通线性模型和广义线性模型的R软件包,支持lasso回归分析。
在这个例子中,我们使用默认的alpha参数值,即1,表示lasso回归。
我们使用交叉验证来选择最优的lambda参数值。
```# 导入glmnet包library(glmnet)# 使用默认的alpha参数值进行lasso回归分析lasso_model <- glmnet(X, Y, alpha = 1)# 使用交叉验证选择最优的lambda参数值cv_model <- cv.glmnet(X, Y, alpha = 1)```结果解读通过运行上述代码,我们得到了lasso回归分析的结果。
我们可以通过以下步骤来解读结果。
1. 可视化lambda和对应的系数收缩路径。

lasso回归筛选变量基因"lasso回归筛选变量基因"——用于基因研究中的变量筛选技术引言:随着高通量技术的发展,基因组数据的获取变得越来越容易。
然而,对于这些大规模数据的分析和挖掘,研究人员面临一个重要的问题:如何从众多的基因中筛选出与所研究现象相关的变量。
lasso回归作为一种变量筛选的统计方法,已经被广泛应用于基因研究领域。
本文将详细介绍lasso 回归在基因研究中的应用过程,逐步回答相关问题。
一、什么是lasso回归?lasso回归(Least Absolute Shrinkage and Selection Operator)是一种融合了正则化和回归分析的统计方法。
它通过对目标变量与相关自变量之间的关系进行建模,从而筛选出与目标变量相关性较强的自变量。
lasso 回归在模型拟合过程中引入了L1正则化项,可以将某些自变量的系数收缩到零,从而实现变量筛选的目的。
二、基因研究中的lasso回归筛选变量在基因研究中,我们常常需要通过分析基因表达数据等信息,来确定哪些基因与某个生理现象或疾病有关。
lasso回归可以帮助我们从海量的基因中筛选出与目标现象相关的变量(基因),以便进一步深入研究。
三、lasso回归流程1. 数据准备:收集相关的基因表达数据或其他基因相关数据,并对数据进行预处理,如去除异常值、标准化等。
2. 构建模型:将所研究的现象(如疾病发生与否)作为目标变量,将基因表达数据等作为自变量,使用lasso回归建立预测模型。
3. 模型训练:使用训练数据集对模型进行训练,通过最小化loss function 来确定模型的系数。
4. 变量筛选:通过调节模型中的正则化参数,使得一些基因的系数为零,即被筛选出来。
这些系数为零的基因即为与目标现象不相关的变量。
5. 模型评估:使用测试数据集对模型进行评估,计算其预测准确率等指标,评估模型的性能。
四、优势与局限性1. 优势:- 变量筛选:通过lasso回归可以从众多的基因中筛选出与目标现象相关的变量,减少研究的复杂性。

基于Lasso的数据特征选择研究近年来,随着数据科学的迅速发展,越来越多的数据被收集并用于各种领域的应用。
然而,大量的数据常常使得数据分析变得困难和耗时。
因此,数据特征选择成为了解决这个问题的关键。
本文将介绍基于Lasso的数据特征选择研究。
一、Lasso回归Lasso是一种用于数据建模和特征选择的线性回归方法。
它不仅可以提高模型的预测能力,还可以选出比较重要的特征。
在普通的线性回归中,我们有如下的模型:$$y=\beta_0+\beta_1x_1+\cdots+\beta_n x_n+\epsilon$$其中,$y$是因变量,$x_1,\cdots,x_n$是自变量,$\epsilon$是误差项,$\beta_0,\cdots,\beta_n$是回归系数。
然而,在实际的应用中,有许多自变量并不对因变量有显著的影响,那么这些自变量的系数应该为0。
Lasso回归通过在普通线性回归中添加一个$L_1$惩罚项来实现这一点,即:$$\text{Minimize }\frac{1}{2}\sum_{i=1}^n(y_i-\beta_0-\sum_{j=1}^n\beta_jx_{ij})^2+\lambda\sum_{j=1}^n|\beta_j|$$其中,$\lambda$是一个调整参数,用于平衡模型的复杂度和预测能力。
Lasso回归具有许多好处,例如可以减少过度拟合、提高模型的稳定性、选择比较重要的特征等。
二、数据特征选择数据特征选择是指从原始的数据集中选择最有价值的特征来构建模型。
常见的数据特征选择方法有Filter方法、Wrapper方法和Embedded方法。
其中,Filter方法是一种基于特征和数据统计学的选择方式,Wrapper方法是一种基于模型的选择方式,而Embedded方法则是将特征选择融入到模型构建过程中。
在所有的特征选择方法中,Embedded方法是最流行的,因为它可以同时提高模型预测和特征选择的准确度。

序号一:概述在数据分析和统计学中,回归分析是一种常用的分析方法,它用于探索变量之间的关系,并进行预测。
而lasson回归则是回归分析的一种方法,它可以用来处理大规模的数据集,并且能够在变量间进行变量选择,避免过拟合的问题。
在本文中,我们将重点介绍如何使用R语言进行lasson回归分析,并且讨论一些常见的数据处理技巧。
序号二:数据预处理在进行lasson回归之前,我们首先需要对数据进行预处理。
这包括数据清洗、缺失值处理、异常值处理等工作。
在R语言中,可以使用一些常用的包如dplyr和tidyverse来进行数据预处理。
在进行数据预处理时,我们需要注意保持数据的完整性和准确性,以确保回归分析的结果具有可靠性。
序号三:模型构建在数据预处理完成之后,我们可以开始构建lasson回归模型。
在R语言中,我们可以使用一些常用的包如glmnet来进行lasson回归分析。
在构建模型时,我们需要注意选择合适的自变量,并且进行交叉验证来选择合适的惩罚参数。
这一步需要结合实际问题和数据特点来进行,以获得最优的模型。
序号四:模型评估在构建好模型之后,我们需要对模型进行评估。
这包括对模型的拟合优度、残差分析、预测能力等方面进行评估。
在R语言中,我们可以使用一些常用的包如caret和rmarkdown来进行模型评估。
通过模型评估,我们可以了解模型的表现如何,并且对模型进行进一步改进和优化。
序号五:结果解释我们需要对lasson回归分析的结果进行解释。
这包括对回归系数的解释、变量选择的意义、预测的可靠性等方面进行解释。
在R语言中,我们可以使用一些常用的包如ggplot2和kableExtra来进行结果可视化和报告呈现。
通过结果解释,我们可以向相关人员、决策者等传达模型的结果和结论,以支持决策和实践。
结论在本文中,我们介绍了如何使用R语言进行lasson回归分析,并且讨论了一些常见的数据处理技巧。
通过对数据预处理、模型构建、模型评估和结果解释的介绍,我们希望读者能够掌握lasson回归分析的基本方法和技巧,并且能够在实际工作中应用和实践。

r语言lasso回归筛选变量代码Lasso回归是一种常见的变量筛选方法,可以用于从众多变量中选择最相关的变量,从而简化模型并提高预测准确率。
在R语言中,可以使用glmnet包实现Lasso回归。
下面介绍如何使用R语言实现Lasso回归筛选变量。
1. 安装和加载glmnet包在R语言中,可以使用install.packages("glmnet")命令安装glmnet 包,然后使用library(glmnet)命令加载glmnet包。
2. 准备数据将要用于Lasso回归的数据加载到R语言中,可以使用read.csv()或read.table()函数加载数据。
在本例中,假设已将数据保存为名为data.csv的CSV文件。
```Rdata <- read.csv("data.csv", header = TRUE)```3. 数据预处理对数据进行预处理以准备进行Lasso回归。
这可能包括删除缺失值、标准化数据等。
```R# 删除缺失值data <- na.omit(data)# 标准化数据data.x <- as.matrix(scale(data[, -1]))data.y <- data[, 1]```4. Lasso回归模型使用glmnet()函数创建Lasso回归模型。
传递自变量矩阵和因变量向量作为参数。
使用cv.glmnet()函数可以执行交叉验证,并计算最佳lambda值。
```R# 创建Lasso回归模型lasso.mod <- glmnet(data.x, data.y, alpha = 1)# 执行交叉验证lasso.cv <- cv.glmnet(data.x, data.y, alpha = 1)```5. 变量选择使用coef()函数可以获得选择的变量和它们的系数。
也可以使用predict()函数预测因变量值。

《套索回归模型在中医临床研究数据的统计应用与R语言实践》篇一摘要:本文将介绍套索回归模型(Lasso Regression Model)在中医临床研究数据的统计应用及其在R语言环境下的实践。
我们将探讨套索回归模型的基本原理、在中医临床数据中的适用性、模型的具体实现以及R语言代码的实践操作。
一、引言随着大数据时代的到来,中医临床研究数据的统计分析和处理变得越来越重要。
套索回归模型作为一种高效的统计工具,能够有效地处理多变量之间的复杂关系,对中医临床数据的分析具有重要价值。
本文将通过理论与实践相结合的方式,展示套索回归模型在中医临床研究数据中的应用。
二、套索回归模型的基本原理套索回归模型是一种用于回归分析的统计方法,它通过引入L1正则化项(即套索项),在回归过程中对系数进行压缩和选择,从而实现对变量选择和降维的目的。
套索回归模型在处理多变量之间复杂关系时,能够有效地避免过拟合和共线性问题,提高模型的预测精度和解释性。
三、套索回归模型在中医临床研究数据的适用性中医临床研究数据通常具有多变量、非线性和高维度的特点,传统的回归分析方法往往难以处理。
而套索回归模型通过引入L1正则化项,能够在保持变量间关系的同时,对不重要的变量进行压缩或剔除,从而实现对数据的降维和简化。
此外,套索回归模型还能够有效地处理共线性问题,提高模型的预测精度和稳定性。
因此,套索回归模型在中医临床研究数据的统计分析中具有广泛的应用前景。
四、套索回归模型的具体实现在R语言环境下,我们可以使用`glmnet`包来实现套索回归模型。
首先,需要安装并加载`glmnet`包。
然后,通过读取中医临床研究数据,进行数据预处理和变量选择。
接着,使用`glmnet`函数进行套索回归模型的训练和参数优化。
最后,利用训练好的模型进行预测和分析。
五、R语言实践操作以下是使用R语言实现套索回归模型的具体步骤:1. 安装并加载`glmnet`包;2. 读取中医临床研究数据,进行数据预处理和变量选择;3. 使用`glmnet`函数进行套索回归模型的训练和参数优化,如设置alpha参数(Lasso的权重)来控制L1正则化的强度;4. 利用训练好的模型进行预测和分析,如计算模型的系数、截距等;5. 绘制模型结果图,如变量系数图、残差图等。

tcga lasso筛选变量实例
TCGA(The Cancer Genome Atlas)是一个旨在理解癌症的分子
结构和基因组学特征的项目,而LASSO(Least Absolute
Shrinkage and Selection Operator)是一种用于特征选择和回归
分析的统计方法。
在TCGA数据中,可以使用LASSO方法来筛选变量,以识别与癌症相关的基因或其他生物标记物。
首先,要使用TCGA数据进行LASSO筛选变量,需要获取适当的TCGA数据集,这可能涉及到访问TCGA数据库或相关的数据存储库。
一旦获取了数据集,就可以使用统计软件(如R或Python中的
scikit-learn库)来实施LASSO算法。
在实施LASSO算法时,需要考虑以下几个步骤:
1. 数据预处理,包括数据清洗、缺失值处理、标准化等。
2. 划分数据集,将数据集分为训练集和测试集,以便评估模型
的性能。
3. 应用LASSO算法,使用训练集对模型进行拟合,通过LASSO
算法选择最相关的变量。
4. 模型评估,使用测试集评估模型的性能,包括模型的预测能
力和所选择变量的有效性。
在TCGA数据中,LASSO算法可以帮助筛选出与癌症相关的基因
或其他生物标记物,这些变量可能有助于理解癌症的发病机制、预
后和治疗反应等方面。
同时,LASSO算法还可以减少模型的复杂性,提高模型的解释性和泛化能力。
总之,使用TCGA数据进行LASSO筛选变量需要经过数据获取、
预处理、模型应用和评估等多个步骤,通过这些步骤可以识别出与
癌症相关的重要变量,为癌症研究和临床实践提供有益信息。

《套索回归模型在中医临床研究数据的统计应用与R语言实践》篇一摘要:随着中医药学的深入发展,大量的临床研究数据亟待通过高效且科学的统计方法进行分析和挖掘。
本文将探讨套索回归模型(Lasso Regression)在中医临床研究数据中的统计应用,并详细介绍如何使用R语言进行实践操作。
一、引言中医临床研究数据具有复杂性和多维性,传统的统计方法往往难以全面、准确地揭示数据间的关系。
套索回归模型作为一种有效的特征选择和降维方法,在处理高维数据时具有独特的优势。
本文将重点介绍套索回归模型在中医临床研究数据中的应用,并详细阐述使用R语言进行实践操作的方法和步骤。
二、套索回归模型概述套索回归模型是一种线性模型,通过在回归系数上添加L1范数约束(即绝对值之和),实现特征选择和降维的目的。
Lasso 回归可以在高维数据中找出对响应变量影响较大的特征,同时对噪声和不重要的特征进行压缩或置零,从而简化模型并提高预测精度。
三、套索回归模型在中医临床研究数据的应用中医临床研究数据通常涉及多个变量,包括患者的年龄、性别、病情、用药等。
这些变量之间可能存在复杂的非线性关系,而套索回归模型能够有效地处理这种高维、非线性的关系。
通过套索回归模型,我们可以找出与疾病发展或治疗效果密切相关的关键变量,为中医临床研究和治疗提供科学依据。
四、R语言实践操作(一)数据准备与预处理首先,我们需要将中医临床研究数据进行整理和清洗,确保数据的准确性和完整性。
然后,根据研究目的和需求,选择合适的变量作为响应变量和特征变量。
(二)R语言安装与包加载在R语言中,我们需要安装并加载必要的包,如`glmnet`(用于实现Lasso回归)和`dplyr`(用于数据处理)。
(三)建立套索回归模型使用`glmnet`包中的函数建立套索回归模型。
通过交叉验证选择合适的惩罚项参数(alpha和lambda),以优化模型的性能。
(四)模型评估与优化使用交叉验证等方法对模型进行评估,通过调整参数或尝试其他方法优化模型性能。

LASS2/TMSG-1基因与肿瘤转移关系的研究进展
左华楚;李秀霞;徐晓艳
【期刊名称】《临床与病理杂志》
【年(卷),期】2014(034)003
【摘要】人源性长寿保障基因2(homo sapiens longevity assurance homologue 2,LASS2)是一个与酵母长寿保障基因l(10ngevity assurance gene1,LAG1)高度同源的人类基因,又被称为肿瘤转移抑制相关基因1(tumor metastasis suppressor gene-1,TMSG-1)。
目前研究发现LASS2/TMSG-1与神经酰胺合成密切相关,同时其与V-ATPase质子泵的相互作用被认为是抑制肿瘤转移的机制之一,多种肿瘤的转移都与此基因的表达相关。
【总页数】6页(P280-285)
【作者】左华楚;李秀霞;徐晓艳
【作者单位】
【正文语种】中文
【中图分类】R737.25
【相关文献】
SS2/TMSG-1基因与肿瘤转移关系的研究进展 [J], 左华楚;李秀霞;徐晓艳
SS2基因对恶性肿瘤转移抑制作用的研究进展 [J], 何俊;王海峰;王剑松
3.MIM及其同源异构体MIM-B基因与肿瘤转移之间关系的研究进展 [J], 陈义刚;丁厚中
4.肿瘤转移抑制相关基因1与肿瘤关系的研究进展 [J], 孙微;李柔;陈晓依;刘博;吕
洋
5.肿瘤转移抑制基因-1与肿瘤关系的研究进展 [J], 杨波;何晓彬
因版权原因,仅展示原文概要,查看原文内容请购买。

㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀∗国家自然科学基金项目(No.61273292)㊁安徽省自然科学基金项目(No.1208085QF122)㊁中央高校基本科研业务费专项资金项目(No.2011HGBZ1329,2011HGQC1013)资助作者简介㊀张靖(通讯作者),女,1987年生,博士研究生,主要研究方向为数据挖掘.E-mail:hfzjwjl@.胡学钢,男,1961年生,教授,博士生导师,主要研究方向为数据挖掘㊁人工智能.李培培,女,1983年生,博士研究生,主要研究方向为数据挖掘.张玉红,女,1979年生,博士,讲师,主要研究方向为数据挖掘.基于迭代Lasso 的肿瘤分类信息基因选择方法研究∗张㊀靖㊀㊀胡学钢㊀㊀李培培㊀㊀张玉红(合肥工业大学计算机与信息学院㊀合肥230009)摘㊀要㊀近年来,基于基因表达谱的肿瘤分类问题引起了广泛关注,为癌症的精确诊断及分型提供了极大的便利.然而,由于基因表达谱数据存在样本数量小㊁维数高㊁噪声大及冗余度高等特点,给深入准确地挖掘基因表达谱中所蕴含的生物医学知识和肿瘤信息基因选择带来了极大困难.文中提出一种基于迭代Lasso 的信息基因选择方法,以获得基因数量少且分类能力较强的信息基因子集.该方法分为两层:第一层采用信噪比指标衡量基因的重要性,以过滤无关基因;第二层采用改进的Lasso 方法进行冗余基因的剔除.实验采用5个公开的肿瘤基因表达谱数据集验证了本文方法的可行性和有效性,与已有的信息基因选择方法相比具有更好的分类性能.关键词㊀基因表达谱,肿瘤分类,迭代Lasso,基因选择中图法分类号㊀TP 391Informative Gene Selection for Tumor Classification Based on Iterative LassoZHANG Jing,HU Xue-Gang,LI Pei-Pei,ZHANG Yu-Hong(School of Computer and Information ,Hefei University of Technology ,Hefei 230009)ABSTRACTTumor classification based on gene expression profiles,which is of tremendous convenience forcancer accurate diagnosis and subtype recognition,has drawn a great attention in recent years.Due to the characteristics of small samples,high dimensionality,much noise and data redundancy for geneexpression profiles,it is difficult to mine biological knowledge from gene expression profiles profoundlyand accurately,and it also brings enormous difficulty to informative gene selection in the tumor classification.Therefore,an iterative Lasso-based approach for gene selection,called Gene Selection Based on Iterative Lasso(GSIL),is proposed to select an informative gene subset with fewer genes and better classification ability.The proposed algorithm mainly involves two steps.In the first step,a gene ranking algorithm,Signal Noise Ratio,is applied to select top-ranked genes as the candidate gene subset,which aims to eliminate irrelevant genes.In the second step,an improved method based on Lasso,Iterative Lasso,is employed to eliminate the redundant genes.The experimental results on 5public datasets validate the feasibility and effectiveness of the proposed algorithm and demonstrate that ithas better classification ability in comparison with other gene selection methods.第27卷㊀第1期㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀模式识别与人工智能㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀Vol.27㊀No.1㊀2014年1月㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀PR &AI㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀Jan.㊀2014Key Words㊀Gene Expression Profile,Tumor Classification,Iterative Lasso,Gene Selection1㊀引㊀言肿瘤目前是威胁人类生命的主要病因之一,预防和治疗肿瘤是全球科学家关注的焦点.随着分子生物科学与信息科学的发展,DNA微阵列(基因芯片)技术因其高通量㊁微型化等特点被广泛应用于疾病诊断㊁临床检验等方面,从而产生大量的基因表达谱数据.DNA微阵列技术不仅有助于对肿瘤进行诊断和分类,而且也为在基因表达水平上研究肿瘤的发生发展机制提供重要依据.自1999年Golub等[1]发表基于基因表达谱分类急性白血病亚型的文章开始,越来越多的研究人员致力于肿瘤分类问题的研究并成功将机器学习模型用于解决该问题[2-5],使其成为DNA微阵列技术应用广㊁发展快的方向之一.然而,肿瘤基因表达谱数据本身通常具有小样本㊁高维数㊁高冗余的特点,从而导致 维数灾难 (Curse of Dimensionality)问题和 过拟合 (Over-fitting)现象[6],使得肿瘤分类问题成为一件有挑战性的工作.为解决上述问题,维数约简(Dimensionality Reduction)方法受到广泛关注,它通过特征抽取(Feature Extraction)或特征选择(Feature Selection)来降低原始空间的维数,两者区别在于,特征抽取指的是对原始特征空间进行某种操作使其投影到一个新的低维特征空间,得到的特征可更好的描述数据,典型的特征抽取方法包括主成分分析㊁线性判别分析等;而特征选择是指从原始特征空间中选取一个最优特征子集以达到降维的目的,通过剔除无关特征和冗余特征,选择的特征子集与分类目标存在高相关性,在其基础上能建立更精确㊁更易理解的模型[7].另一方面,肿瘤基因表达谱数据集中仅有少量基因与疾病密切相关,称为信息基因(Informative Genes)[8],因而可见特征选择比特征抽取具有优势,更适用于肿瘤分类问题研究.信息基因选择(即特征选择)已逐步成为肿瘤分类问题的核心研究内容.信息基因选择方法中最简单常用的一类方法是排序法,这类方法通常采用某种指标对所有基因 打分 (分数反映了各个基因对分类目标的重要性程度),然后根据分数高低对基因排序,最后选择Top K个基因作为信息基因(K一般取50~200)[9].目前常用的排序法包括信噪比指标(Signal Noise Ratio,SNR)[1],t-检验(t-statistic)[9],信息增益(Information Gain,IG)[9]等,其中,信噪比指标是应用最为广泛的.排序法不依赖于具体的分类算法,通过该方法得到的信息基因子集可有效避免 过拟合 现象,同时由于该方法具有实现简单和时间复杂度低等优点,因而较适用于高维的基因表达谱数据.一般情况下,基因排序法选择的基因通常是高相关的[10].这种相关性主要是由于如果一个基因排名较高,排序法极有可能选择与之高相关的其它基因作为信息基因,从而导致冗余.过多的冗余基因将使得到的基因子集规模变大,而判别能力下降;同时,特征冗余作为额外的计算负担,会使结果倾斜易导致错误分类.为有效去除冗余基因,学者们提出一些解决方法.Tan等[11]首先使用不同的基因排序方法构造候选基因子集,然后结合遗传算法与支持向量机分类器来去除冗余;李颖新等[12]采用两两冗余分析及基于支持向量机分类模型的灵敏度分析法进行冗余基因的剔除;王树林等[13]提出一种以支持向量机分类性能为评估准则的寻找信息基因的启发式宽度优先搜索算法来剔除冗余基因;Chuang等[14]结合二进制粒子群优化算法和遗传算法,使用K近邻分类器来减少冗余基因.上述方法在一定程度上解决了冗余基因带来的负面影响,但由于在基因选择过程直接利用学习算法来评估基因子集,分类模型与基因选择之间相互依赖,存在较高风险的过拟合㊁泛化性能差等问题,同时无法保证得到稀疏解,且构建分类模型需大量的计算开销.因此,设计鲁棒高效的信息基因选择算法是肿瘤分类领域的研究重点. Lasso方法因其高效的性能在特征选择领域引起广泛关注[15-16].Lasso方法是一种基于线性回归模型的降维方法,它克服传统方法在选择模型上的不足,通过l1范式约束可使模型得到稀疏解[17],在基因表达谱分析中被广泛应用.但考虑若基因之间相关性较强,且互为冗余,Lasso方法极有可能将这组基因误认为是信息基因;同时由于Lasso具有凸优化的性质,使其在构建线性回归模型时过于严格,要求样本数据与模型完全拟合,会出现过拟合问题.因此本文在Lasso方法上进行改进,提出迭代Lasso (Iterative Lasso,ILasso)方法来去除冗余基因,解决 过拟合 问题.鉴于肿瘤基因表达谱数据本身的特点,本文研究目标是采用尽可能少的信息基因以获得尽可能高05模式识别与人工智能㊀㊀㊀27卷的样本分类准确率.本文提出一种新的肿瘤分类信息基因选择方法,即基于ILasso的信息基因选择方法(Gene Selection Based on Iterative Lasso,GSIL).该方法分为两层:第一层采用信噪比指标衡量基因的重要性,以过滤无关基因;第二层采用改进的Las-so方法进行冗余基因的剔除.实验采用5个公开的肿瘤基因表达谱数据集作为具体的研究对象,利用本文方法得到基因数量少且分类能力较强的信息基因子集,并与已有的信息基因选择方法进行分析比较以验证所设计方法的可行性和有效性.2㊀GSIL方法系统框架针对肿瘤基因表达谱的信息基因选取,我们提出基于ILasso的信息基因选择方法GSIL,该方法基于信噪比指标和改进的Lasso方法,能有效过滤无关基因并剔除冗余基因.图1给出基于ILasso的信息基因选择方法GSIL的系统框架.GSIL特征选择方法分为两层:第一层采用信噪比方法SNR过滤不相关基因;第二层使用改进的Lasso方法剔除冗余基因.假设数据(X,Y)包含n个样本,m个基因,GSIL 方法的具体步骤如下:step1㊀信噪比方法过滤不相关基因,从m个基因中选择得分较高的mᶄ个基因;step2㊀改进Lasso方法剔除冗余基因,对mᶄ个基因进行特征选择,得到mᵡ个信息基因; step3㊀建立分类模型并评价.图1㊀基于ILasso的信息基因选择方法的系统框架Fig.1㊀Framework of GSIL for informative gene selectionstep1和step2的具体实现将在第3节和第4节中详细阐述.step3中为公平评价基因子集的分类性能,实验采用多种不同的学习算法来验证,并采用留一交叉验证(Leave-One-Out Cross Validation, LOOCV)方法作为评估标准.LOOCV方法是交叉验证方法的一个特例,它每次选择一个样本作为测试样本,再用样本集中的剩余样本建立分类模型,当所有样本都被测试一遍后,记录分类精度(Accuracy).因此,经过基因选择步骤后,本文使用下列4种分类器来评价本文提出的信息基因选择算法GSIL的有效性:支持向量机(SVM)㊁K近邻(KNN)㊁决策树C4.5和随机森林Random Forest,并采用LOOCV作为分类精度的评价方法.3㊀基于信噪比指标过滤无关基因鉴于肿瘤基因数据高维小样本的特点,在分类前需采用各种方法对基因表达谱数据进行降维和剔除冗余基因等处理,从而最大限度的提高肿瘤样本的分类性能.一般而言,基因表达谱数据的维数为几千㊁甚至上万,而在如此高维的数据中,与某一疾病相关的基因数目大多为几十个,基本不会超过一百个,因此基因表达谱数据中存在大量的无关基因,这些基因在不同组织样本中的表达值差异度很小,对于分类无法提供有用的信息,还会导致特征空间的增大,为信息基因选择带来极大困难.基因排序法是过滤无关基因的一类常用方法,其过程通常采用某种指标对所有基因 打分 ,然后根据分数高低将排名靠后的基因删除,选择分数高的若干基因作为信息基因.由于排序法独立于具体的分类算法,因此适用于信息基因的初步选择,生成候选基因子集.信噪比评价指标在排序法中是最为简单常用的一种方法.在进行基因选择过程中,首先采用简单高效的信噪比指标在原始特征空间过滤无关基因,选择出与类别属性相关性较高的基因.针对每个基因,通过㊀㊀㊀SNR(g i)=μ+(g i)-μ-(g i)σ+(g i)+σ-(g i).(1)来衡量其重要性,信噪比值越大,基因的重要性越高.其中,μ+(g i)和μ-(g i)分别表示第i个基因g i在正类和负类中的平均表达值,而σ+(g i)和σ-(g i)分别为第i个基因g i在两类中的标准差.为直观表示,以实验部分的结肠癌Colon数据集为例,该数据集包括62个样本,2000个基因.图2给出该数据集全部基因的信噪比值大小及其相应的区间分布.由图2可知,大部分基因的信噪比值在该数据集上都较小,如有1000多个基因的信噪比值均低于0.2,表明这些基因难以区分类别,可作为无关基因处理,只有为数不多的基因才与分类目标密切相关.151期㊀㊀㊀㊀张㊀靖㊀等:基于迭代Lasso的肿瘤分类信息基因选择方法研究图2㊀Colon数据集的信噪比值和区间分布图Fig.2㊀Signal-noise ratio and distribution of Colon dataset 过滤无关基因的具体方法是首先以式(1)为m个基因分别计算其信噪比值,然后按照信噪比值的大小对全部基因进行降序排列,并选择前mᶄ个基因作为候选基因子集,通常mᶄ<<m.然而,这些选定的mᶄ个基因之间往往是高相关的,从而导致冗余.这是由于基因在细胞中存在共表达现象,即如果一个基因排名较高,排序方法极有可能选择与之高相关的其它基因作为信息基因,过多的冗余基因将使得到的候选基因子集规模变大,而判别能力下降[18];同时,特征冗余作为额外的计算负担,会使结果倾斜从而导致错误分类,不利于从生物学角度理解基因表达数据,因此,本文采用改进的Lasso方法进一步剔除冗余基因,从mᶄ个基因中得到mᵡ个信息基因,以获得最优信息基因子集.4㊀基于Lasso方法剔除冗余基因Lasso方法因其高效的性能在特征选择领域引起广泛关注.Lasso方法是一种基于线性回归模型的降维方法,克服了传统方法在选择模型上的不足,通过l1范式约束可使模型得到稀疏解,在基因表达谱分析中被广泛应用.但经过信噪比指标过滤无关基因后所得到的信息基因之间通常是高相关的,如果一组基因之间相关性较强且互为冗余,直接使用Lasso方法处理极有可能将这组基因都误认为是信息基因;同时,由于Lasso具有凸优化的性质,使其在构建线性回归模型时过于严格,要求样本数据与模型完全拟合,会出现 过拟合 问题.张靖等[19]提出一种基于Lasso的信息基因选择算法 K-split Lasso,其基本思想是将训练集随机均分为K份,使用Lasso方法对每份子集进行特征选择,并将选择出来的所有基因子集合并,重新进行特征选择,最终得到一个基因子集.K-split Lasso方法可减少计算开销,提高模型的分类精度,在一定程度上解决 过拟合 问题.然而,K-split Lasso方法具有以下两个缺陷.首先,K-split Lasso方法在设计过程中是随机划分基因集合为K份,并将划分后的K 个基因子集作为独立的个体处理,没有考虑各个子集内部基因的关联性,易将冗余基因作为信息基因保留下来,同时随机划分也增加运行结果不稳定性的风险.其次,K-split Lasso方法在选择信息基因之前未考虑无关基因和噪声基因对基因选择的负面影响,从而增加算法的负担,无法有效剔除冗余基因.因此,为有效剔除候选基因子集中的冗余基因,解决K-split Lasso方法的缺陷,本文采取序列前向搜索策略,并结合迭代优化的思想,提出一种改进的Lasso方法,称之为ILasso方法.最小绝对收缩和选择算子(Least Absolute Shrinkage and Selection Operator,Lasso)[20]是一种收缩估计方法.通过构造一个惩罚函数得到较为简练的模型,收缩变量的系数.一些相关度较低的变量系数被压缩为0,同时达到变量选择及参数估计的目的,保留子集收缩的优点是一种有效处理具有复共线性数据的有偏估计.张靖等[19]对Lasso方法做了详细介绍,并且介绍一种快速近似求解Lasso凸优化问题的算法,最小角回归算法(Least Angle Regression,LARS)[21].本文与文献[19]类似,在实验过程中采用LARS算法选择信息基因,文献[19]中2.1节给出LARS算法的具体实现流程.通过采取序列前向搜索策略,并结合迭代优化的思想,采用ILasso方法剔除冗余基因.算法的主要思想是通过划分候选基因子集降低每次处理的基因维数,弱化基因之间的相关性,迭代使用LARS算法将选择的信息基因添加到当前已选的信息基因子集中.迭代的好处是在每次使用LARS算法选择信息基因的过程中都选择出与分类任务最为相关的基因,减少冗余基因的干扰,可较大程度剔除冗余基因.具体步骤见算法1.在算法1的描述中,假设ILasso算法的输入为过滤无关基因后的候选训练集合(X,Y),包含n个样本,mᶄ个基因{G1,G2, ,G mᶄ},X(G1,G2, ,G mᶄ)是mᶄ个n维的自变量,Y=(Y1,Y2, ,Y n)T是n维响应变量,参数K为划分的基因子集份数.输出是mᵡ个信息基因,即最优信息基因子集S best.首先设最优信息基因子集S best为空集,将经过信噪比排序后的基因列表G list平均划分为K个基因25模式识别与人工智能㊀㊀㊀27卷子集,设G list(i)是将基因集划分为K份后的第i (1ɤiɤK)份基因子集;然后对第1份基因子集G list(1)利用LARS算法进行变量系数压缩的计算,选择系数不为0的基因加入到S best中,并将S best与第2份基因子集G list(2)合并作为当前的基因子集,重新采用LARS算法进行基因选择,删除系数压缩为0的基因,更新当前的信息基因子集S best,依次迭代到第K份G list(K),最终得到的信息基因子集即为最优信息基因子集S best.算法1 ILasso算法输入㊀(X,Y)//过滤无关基因后的候选训练集合G list={G1,G2, ,G mᶄ}//基因列表K//划分的子集个数输出㊀S best//最优信息基因子集Begin㊀X进行标准化处理(均值为0,方差为1);Y进行中心化处理(均值为0);For i=1to mᶄ㊀对G list中每个基因G i计算其与类别Y的信噪比值; End根据信噪比值的大小对G list中的基因进行升序排序;S best=NULL;㊀㊀//初始化最优信息基因子集S best为空For i=1to K㊀G list(i)={G(i-1)mᶄ/K+1, ,G i mᶄ/K};㊀//G list(i)是第i份基因列表EndFor i=1to K㊀G list(i)=S bestɣG list(i);//将当前信息基因子集S best添加到G list(i)中㊀X i=X(G list(i));//X i为G list(i)基因列表对应的数据子集㊀S best=LARS(X i,Y);//利用LARS算法更新信息基因子集S best,最终S best=mᵡEndEnd这里需要说明的是,在使用信噪比指标过滤无关基因的步骤中所得到的候选信息基因是按照得分由高到低进行排序,即重要性越高的基因排名越靠前;而在采用ILasso方法剔除冗余基因的步骤中,将候选信息基因反向排序(为便于算法描述,在算法1中重新计算每个基因与类别Y的信噪比值,并根据值的大小对基因进行升序排序),然后划分基因集合,即首先选取重要性较低的基因采用LARS算法进行基因选择.这样做的目的是考虑到信噪比指标得分高的基因更能反映类别特征,其重要性程度也越高,升序排序在剔除冗余基因的过程中更利于保留与分类任务高相关的基因,最大程度地剔除冗余基因并减少误删信息基因的可能性.图3㊀ILasso算法流程Fig.3㊀Flowchart of ILasso algorithm图3给出ILasso方法的具体实现流程.(X,Y)是过滤无关基因后的候选训练集合,将基因列表G list的基因按照信噪比值由小到大进行排序,并依次生成K个基因子集,记为G list(i)(1ɤiɤK),目标是得到最优信息基因子集S best.ILasso算法首先初始化S best为空集,然后依次将当前的信息基因子集S best添加到G list(i)中作为新的基因子集,基于更新后的G list(i)基因列表得到其对应的数据子集X i,迭代采用LARS算法剔除冗余基因,保留信息基因,最后经过K次迭代后得到的信息基因集合就是最优信息基因子集S best.从理论上讲,Lasso方法本就是一种较好的特征选择方法,它能将与类标签强关联的变量选择出来, ILasso方法是在其基础上引入迭代优化的思想,通过调整参数K的取值进一步剔除冗余特征,解决 过拟合 问题,同时保留与类标签强相关的变量.针对本文的肿瘤基因表达谱数据集,假设G i㊁G j均是与分类任务强相关的信息基因,且G i(G j)为G j(G i)的冗余基因,若直接采用其它基因选择方法处理,G i㊁G j有可能均作为信息基因被保留下来,而使用GSIL 方法,假设在第一层过滤无关基因的过程中G i和G j 都作为信息基因被选择,然而在第二层使用ILasso 方法进行二次选择时,因为算法采用的是迭代策略,同一基因可能会被多次选择,所以不论G i㊁G j原先是否被划分到同一基因子集中,都将对新一轮选择出的基因迭代进行基因选择,在多次选择之后,冗余基因可被有效剔除.由上可知,ILasso方法不仅适用于高维小样本数据的基因选择,能有效剔除冗余基因,同时由于Lasso方法本身具有得到稀疏解的性能,也使得选择出的信息基因个数较少.因此,ILasso方法是一种有效的基因选择方法.351期㊀㊀㊀㊀张㊀靖㊀等:基于迭代Lasso的肿瘤分类信息基因选择方法研究5㊀实验与结果分析5.1㊀实验数据和实验环境本文的分析对象为5个公开的基因表达谱数据集,包括Colon㊁Prostate㊁Lymphoma㊁Leukemia和Lung,这些数据集均可从BRB-ArrayTools[22]主页下载(/~brb/DataArchive_ New.html).BRB-ArrayTools是一款为DNA基因芯片数据分析而设计的集成软件包,由Dr.Richard Simon所领导的生物识别小组所开发.具体数据集描述见表1.表1㊀实验数据集描述Table1㊀Description for experimental datasets数据集序号数据集名称基因数量样本数量(正类/负类)类别数1Colon200062(40/22)22Prostate12600102(52/50)23Lymphoma712977(58/19)24Leukemia712972(25/47)25Lung12533181(31/150)2 1)结肠癌数据集Colon[23].该数据集共包含62例样本,其中40例为结肠癌(Colon Cancer,CC)组织样本㊁22例为正常(Normal,N)组织样本,每例样本由2000个基因表达谱组成.2)前列腺癌数据集Prostate[24].该数据集共有102例样本,包括52例前列腺癌(Prostate Cancer, PC)组织样本和50例正常(Normal,N)组织样本,每例样本由12600个基因表达谱组成.3)淋巴癌数据集Lymphoma[25].该数据集共包含77例样本,其中58例为弥漫性大B细胞淋巴瘤(Diffuse Large B-Cell Lymphoma,DLBCL)样本㊁19例为滤泡性淋巴瘤(Follicular Lymphoma,FL)样本,每例样本由7129个基因表达谱组成.4)急性白血病数据集Leukemia[1].该数据集共含72例样本,包括25例急性髓细胞白血病(AML)样本和47例急性淋巴细胞白血病(ALL)样本,每例样本由7129个基因表达谱组成.5)肺癌数据集Lung[26].该数据集共含有181例样本,包括31例恶性胸膜间皮瘤(Malignant Pleu-ral Mesothelioma,MPM)样本和150例恶性胸腺癌(ADeno CArcinoma,ADCA)样本,每例样本由12533个基因表达谱组成.需要说明的是通过观察数据发现,Prostate和Lung这两个数据集中部分基因列的值全为零,这些值全为零的基因列不仅对基因选择任务没有任何帮助,还有可能影响基因选择的结果,因此剔除了Prostate中的394列噪音基因数据和Lung中的121列噪音基因数据,得到的Prostate数据集的基因数量由原始的12600降为12206,Lung数据集的基因数量由原始的12533降为12412.本文采用的实验环境配置:Intel Xeon5110双核处理器,2GB内存的PC机,Windows XP的操作系统,Weka3.7.3+Matlab7.0的开发环境.使用We-ka[27]工具进行分类模型构建和LOOCV性能评价,在实验部分使用的4种分类器,即SVM㊁KNN㊁决策树C4.5和随机森林Random Forest,都集成在Weka 软件中.实验过程中对各算法的重要参数设置如下: SVM的核函数kernel设置为多项式核函数(Polyno-mial Kernel Function),KNN的邻居个数K设置为10,C4.5的用于修剪的置信因子(confidenceFactor)设置为0.25,Random Forest的生成树的个数(numTrees)设置为10.5.2㊀实验结果分析本文设计的算法GSIL分为两层:第一层按照信噪比值的大小对全部m个基因进行降序排列,并选择前mᶄ个基因作为候选基因子集,通常mᶄ<<m,从而有效降低基因维数,去除无关基因;第二层在候选基因子集的基础上采用ILasso方法进一步剔除冗余基因,从mᶄ个基因中得到mᵡ个信息基因,以获得最优信息基因子集.首先,本文对第一层的实验结果分析.通常情况下,信噪比方法根据基因得分的高低选择Top50~ 200个基因.本文初步选择Top100(mᶄ=100)个基因作为候选基因子集,为检验候选基因子集的性能,本文以5个实验数据集通过信噪比方法选出的Top 100个基因作为样本的信息基因,采用4种不同的分类器分别构建模型,取分类精度平均值作为最终的分类精度.表2比较原始数据集和经过信噪比方法选择后的候选基因子集的基因个数和分类性能,表格中的粗体值表示当前方法的分类准确率最高.由表2可知,经过信噪比方法过滤后的候选基因子集的分类精度明显高于原始基因集合的分类精度,例如Lung数据集的分类精度达到98.62%,这说明本实验保留的都是与分类任务较为关联的特征,过滤的大多基因都是无关基因.通过去除大量的无关基因,利用选出的Top100个基因即可实现对原始样本集较为精确的分类,包含了原始样本集的完整分类信息.45模式识别与人工智能㊀㊀㊀27卷。

R语⾔实现LASSO回归的⽅法Lasso回归⼜称为套索回归,是Robert Tibshirani于1996年提出的⼀种新的变量选择技术。
Lasso是⼀种收缩估计⽅法,其基本思想是在回归系数的绝对值之和⼩于⼀个常数的约束条件下,使残差平⽅和最⼩化,从⽽能够产⽣某些严格等于0的回归系数,进⼀步得到可以解释的模型。
R语⾔中有多个包可以实现Lasso回归,这⾥使⽤lars包实现。
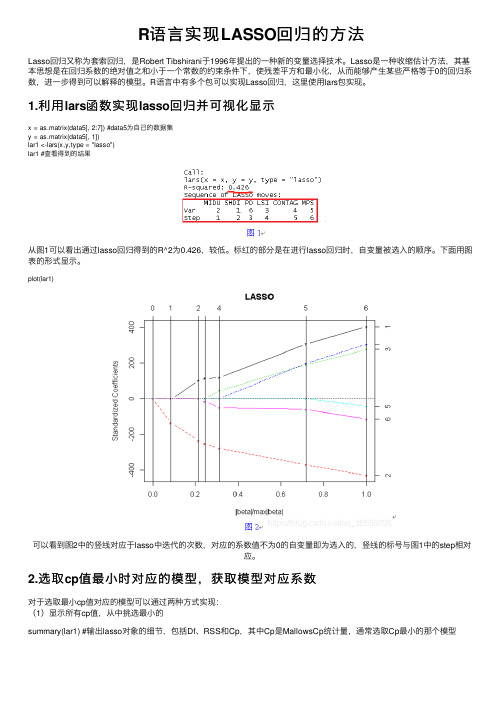
1.利⽤lars函数实现lasso回归并可视化显⽰x = as.matrix(data5[, 2:7]) #data5为⾃⼰的数据集y = as.matrix(data5[, 1])lar1 <-lars(x,y,type = "lasso")lar1 #查看得到的结果从图1可以看出通过lasso回归得到的R^2为0.426,较低。
标红的部分是在进⾏lasso回归时,⾃变量被选⼊的顺序。
下⾯⽤图表的形式显⽰。
plot(lar1)可以看到图2中的竖线对应于lasso中迭代的次数,对应的系数值不为0的⾃变量即为选⼊的,竖线的标号与图1中的step相对应。
2.选取cp值最⼩时对应的模型,获取模型对应系数对于选取最⼩cp值对应的模型可以通过两种⽅式实现:(1)显⽰所有cp值,从中挑选最⼩的summary(lar1) #输出lasso对象的细节,包括Df、RSS和Cp,其中Cp是MallowsCp统计量,通常选取Cp最⼩的那个模型图3显⽰了lasso回归中所有的cp值,选择最⼩的,即上图标红的部分,对应的df=3,最前⾯⼀列对应迭代次数(即步数),step=2 。
(2)直接选取最⼩的cp值lar1$Cp[which.min(lar$Cp)] #选择最⼩Cp,结果如下:与图3中标红的部分结果⼀样,但是要注意,2表⽰的是step⼤⼩。
3.选取cp值最⼩时对应的模型系数(1)获取所有迭代系数,根据step⼤⼩选择cp值最⼩对应的⾃变量系数值lar1$beta #可以得到每⼀步对应的⾃变量对应的系数图4标红的部分就是step=2对应的cp值最⼩时对应的模型的⾃变量的系数(2)获取指定迭代次数(即步数)对应的⾃变量的系数,可以通过下⾯的代码实现:coef <rs(lar,mode="step",s=3) #s为step+1,也⽐图2中竖线为2的迭代次数对应,与图3中df值相等;s取值范围1-7.coef[coef!=0] #获取系数值不为零的⾃变量对应的系数值与图4中标红部分⼀样。

基于多组学Lasso回归分析构建肝癌预测模型罗焱瑞;赵倩【期刊名称】《天津医科大学学报》【年(卷),期】2024(30)3【摘要】目的:通过生物信息学方法整合肝癌转录组和甲基化组数据,利用Lasso回归分析筛选肝癌特异性标志物,并构建肿瘤预测模型。
方法:在GEO(gene expression omnibus)中下载GSE70091、GSE77314数据集,共53例肝癌患者的转录组测序(RNA-seq)和全基因组甲基化测序(WGBS)数据,分别将肝癌与癌旁对照间的转录组、甲基化组数据进行差异分析,并对差异表达基因(DEG)和差异甲基化基因(DMG)进行整合,以筛选出候选肝癌标志基因。
对候选肝癌标志基因进行GO(gene ontology)和Reactome通路富集分析,使用Lasso回归分析筛选标志基因,构建肝癌预测模型,并在其他队列中进行性能验证。
结果:共筛选出288个DEG(|log2FC|>1,P.adj<0.05)和28528个DMG(P<0.05),通过DEG和DMG的交叉分析找到51个高甲基化下调(Hyper-Down)基因和111个低甲基化上调(Hypo-Up)基因。
GO和Reactome通路富集分析显示,Hypo-Up基因主要富集在细胞有丝分裂通路上(FDR<0.05,P<0.05),Hyper-Down基因主要与转录激活的功能相关(FDR<0.05,P<0.05)。
使用Lasso回归分析筛选出11个具有非零系数的基因并构建肝癌预测模型。
最后在GSE77314、外部验证队列(TCGA-LIHC)中验证出模型曲线下面积(AUC)分别为1、0.998。
结论:通过整合肝癌多组学数据,使用Lasso回归分析筛选出11个基因标志物,并构建肝癌预测模型。
【总页数】8页(P205-211)【作者】罗焱瑞;赵倩【作者单位】天津医科大学基础医学院细胞生物学系【正文语种】中文【中图分类】R735.7【相关文献】1.基于多组学数据的乳腺癌预后预测模型构建2.基于Lasso回归构建生物标志物影响代谢综合征的风险预测模型3.基于LASSO回归的活动性肺结核列线图预测模型的构建及验证4.基于LASSO-Logistic回归构建胸腔镜术后患者生活质量影响因素的预测模型5.基于CT影像组学及临床多因素回归分析预测肝细胞肝癌TACE术后中重度腹痛的模型构建因版权原因,仅展示原文概要,查看原文内容请购买。

基于LASSO回归筛选影响肺腺癌患者预后的糖酵解相关基因杜也;米热阿依·阿布都热孜克;左冉;袁东琪;霍庚崴;陈金良;张翠翠;孟昭婷;陈鹏【期刊名称】《中国肿瘤临床》【年(卷),期】2023(50)1【摘要】目的:确定用于评估肺腺癌(lung adenocarcinoma,LUAD)患者糖酵解相关基因的风险评分模型。
方法:使用公共数据库癌症基因组图谱(The Cancer Genome Atlas,TCGA)中LUAD患者转录组数据,通过基因富集分析(gene set enrichment analysis,GSEA)、差异表达基因(differentially expressed genes,DEGs)分析和最小绝对收缩选择算子(least absolute shrinkage and selection operator,LASSO)回归分析构建风险预测模型。
通过Kaplan-Meier分析、受试者工作特征(receiver operating characteristic,ROC)曲线、单因素及多因素Cox回归分析验证模型预测性能。
使用CIBERSORT算法计算高、低风险两组免疫细胞浸润差异。
构建用于临床预测患者预后的列线图。
结果:识别出3个糖酵解相关基因集,筛选出6个糖酵解相关基因构建风险评分模型。
高风险组总生存率显著低于低风险组,验证性结果显示该模型有良好的预测性能。
高、低风险两组的免疫细胞浸润情况存在显著差异。
列线图的构建开发了一种可以预测LUAD患者生存率的定量方法。
结论:基于糖酵解相关基因构建的风险评分模型为早期LUAD患者预测预后提供了新型生物标志物。
【总页数】6页(P16-21)【作者】杜也;米热阿依·阿布都热孜克;左冉;袁东琪;霍庚崴;陈金良;张翠翠;孟昭婷;陈鹏【作者单位】天津医科大学肿瘤医院肺部肿瘤内科【正文语种】中文【中图分类】R73【相关文献】1.基于生物信息学分析的肺腺癌诊断及预后相关基因筛选2.转移性乳腺癌患者预后相关关键基因筛选、预后预测模型构建及验证3.糖酵解相关基因TXN、MET、KIF20A对胰腺癌患者预后评估的意义4.通过生物信息学分析筛选影响胃腺癌患者预后的铁死亡相关基因5.基于生物信息学预测甲状腺癌预后相关高风险糖酵解基因及其与患者预后的关系因版权原因,仅展示原文概要,查看原文内容请购买。

用R进行Lassoregression回归分析glmnet是由斯坦福大学的统计学家们开发的一款R包,用于在传统的广义线性回归模型的基础上添加正则项,以有效解决过拟合的问题,支持线性回归,逻辑回归,泊松回归,cox回归等多种回归模型,链接如下/web/packages/glmnet/index.html对于正则化,提供了以下3种正则化的方式1.ridge regression,岭回归sso regression,套索回归3.elastic-net regression,弹性网络回归这3者的区别就在于正则化的不同,套索回归使用回归系数的绝对值之和作为正则项,即L1范式;岭回归采用的是回归系数的平方和,即L2范式;弹性网络回归同时采用了以上两种策略,其正则项示意如下可以看到,加号左边对应的是lasso回归的正则项,加号右边对应的是ridge回归的正则项。
在glmnet中,引入一个新的变量α, 来表示以上公式可以看到,随着α取值的变化,正则项的公式也随之变化1.alpha = 1, lasso regression2.alpha = 0, ridge regression3.alpha 位于0到1之间, elastic net regression利用alpha的取值来确定回归分析的类型,然后就只需要关注lambda值就可以了。
除此之外,还有一个关键的参数family, 用来表示回归模型的类型,其实就是因变量y的数据分布,有以下几种取值1.gaussian, 说明因变量为连续型变量,服从高斯分布,即正态分布,对于的模型为线性回归模型2.binomial, 说明因变量为二分类变量,服从二项分布,对应的模型为逻辑回归模型3.poisson, 说明因变量为非负正整数,离散型变量,服从泊松分布,对应的模型为泊松回归模型4.cox, 说明因变量为生存分析中的因变量,同时拥有时间和状态两种属性,对应的模型为cox回归模型5.mbinomial, 说明因变量为多分类的离散型变量,对应的模型为逻辑回归模型6.mgaussian, 说明因变量为服从高斯分布的连续型变量,但是有多个因变量,输入的因变量为一个矩阵,对应的模型为线性回归模型理解这两个参数之后,就可以使用这个R包来进行分析了。

lasso用法范文拉索(Lasso)是一种统计学上常用的回归分析方法,也是机器学习中的一个重要技术。
它在线性回归的基础上进行了一定的改进,可以用于特征选择、参数估计和模型预测等任务。
本文将详细介绍Lasso的用法,并探讨它在实际应用中的一些注意事项。
1. Lasso回归模型Lasso(Least Absolute Shrinkage and Selection Operator)回归模型是一种利用L1正则化进行特征选择的线性回归模型。
其目标函数可以写作:L(β) = 1/2n ∑(yi - Xiβ)² + λ∑,β其中,n是样本数量,yi是第i个样本的实际观测值,Xi是该样本对应的特征向量,β是回归系数,λ是正则化参数。
2.特征选择Lasso回归通过引入L1正则化项,使得回归系数β中的一些分量可以被压缩到零,从而实现特征选择。
具体而言,当λ趋近于零时,Lasso 回归与普通的线性回归模型相同,不会剔除任何特征。
当λ增大时,一些特征的回归系数会收缩到零,这些特征可以被认为是无关变量,不对模型预测起作用。
特征选择的优势在于可以减小模型的复杂度,提高模型的泛化能力,同时可以降低噪声特征对模型的影响,提高模型的稳定性。
3.参数估计L1正则化使得Lasso回归的优化问题更加复杂,采用传统的梯度下降等方法难以求解。
常用的解法是利用坐标下降算法(Coordinate Descent)或最小角回归(Least Angle Regression,LAR)来求解。
坐标下降算法的基本思想是固定其他回归系数,通过最小化目标函数对当前回归系数的偏导数来更新当前回归系数。
这个过程通过迭代进行,直到收敛。
LAR算法是一种改进的坐标下降算法,它在每一次迭代中选择紧邻当前残差的变量进行更新。
这样做的好处是可以减少迭代的次数,加快算法的收敛速度。
4.模型预测Lasso回归模型可以用于预测未知样本的响应变量。
当模型训练完成后,给定一个新样本的特征向量,通过与回归系数的相乘求和得到预测值。

lasso-var用法摘要:一、lasso-var 简介sso-var 的定义sso-var 的作用二、lasso-var 的用法sso-var 的基本语法sso-var 的参数说明sso-var 的返回值三、lasso-var 的实际应用1.示例代码2.代码解析四、lasso-var 的优缺点分析1.优点2.缺点五、总结sso-var 的重要性和应用场景2.对未来lasso-var 的发展展望正文:lasso-var 是R 语言中一个非常重要的函数,它用于解决变量选择问题。
通过lasso-var,我们可以找到一组最优的变量,这些变量对于预测目标变量具有较高的准确性。
lasso-var 是基于L1 范数正则化的方法,通过引入正则项,使得模型在训练过程中选择更少的变量,从而提高模型的泛化能力。
一、lasso-var 简介lasso-var 是R 语言中的一个包,主要用于变量选择。
它可以应用于线性回归、逻辑回归、泊松回归等多种模型中,帮助我们找到一个最佳变量集合。
二、lasso-var 的用法要使用lasso-var,首先需要安装并加载该包。
在R 中输入以下命令:```Rinstall.packages("lasso")library(lasso)```接下来,我们可以使用lasso-var 函数进行变量选择。
假设我们有一组数据,其中x 为输入变量,y 为目标变量,可以采用以下代码:```Rlasso_var <- lasso.var(y ~ x, data = data_frame)```其中,`data_frame`为数据框,y 和x 分别为目标变量和输入变量。
三、lasso-var 的实际应用为了更好地理解lasso-var 的实际应用,我们可以通过一个示例来说明。
假设我们有一组鸢尾花的数据,包含4 个输入变量(花萼长度、花萼宽度、花瓣长度、花瓣宽度)和1 个目标变量(种类)。