具有遗传性疾病和性状的遗传位点分析
- 格式:doc
- 大小:877.00 KB
- 文档页数:23

中国研究生数学建模竞赛历届竞赛题目第一届2004年题目A题发现黄球并定位B题实用下料问题C题售后服务数据的运用D题研究生录取问题第二届2005年题目A题HighwayTravelingtimeEstimateandOptimalRoutingB题空中加油C题城市交通管理中的出租车规划D题仓库容量有限条件下的随机存贮管理第三届2006年题目A题AdHoc网络中的区域划分和资源分配问题B题确定高精度参数问题C题维修线性流量阀时的内筒设计问题D题学生面试问题第四届2007年题目A题建立食品卫生安全保障体系数学模型及改进模型的若干理论问题B题械臂运动路径设计问题C题探讨提高高速公路路面质量的改进方案D题邮政运输网络中的邮路规划和邮车调运第五届2008年题目A题汶川地震中唐家山堪塞湖泄洪问题B题城市道路交通信号实时控制问题C题货运列车的编组调度问题D题中央空调系统节能设计问题第六届2009年题目A题我国就业人数或城镇登记失业率的数学建模B题枪弹头痕迹自动比对方法的研究C题多传感器数据融合与航迹预测D题110警车配置及巡逻方案第七届2010年题目A题确定肿瘤的重要基因信息B题与封堵渍口有关的重物落水后运动过程的数学建模C题神经元的形态分类和识别D题特殊工件磨削加工的数学建模第八届2011年题目A题基于光的波粒二象性一种猜想的数学仿真B题吸波材料与微波暗室问题的数学建模C题小麦发育后期茎轩抗倒性的数学模型D题房地产行业的数学建模第九届2012年题目A题基因识别问题及其算法实现B题基于卫星无源探测的空间飞行器主动段轨道估计与误差分析C题有杆抽油系统的数学建模及诊断D题基于卫星云图的风矢场(云导风)度量模型与算法探讨第十届2013年题目A题变循环发动机部件法建模及优化B题功率放大器非线性特性及预失真建模C题微蜂窝环境中无线接收信号的特性分析D题空气中PM2.5问题的研究attachmentE题中等收入定位与人口度量模型研究F题可持续的中国城乡居民养老保险体系的数学模型研究第十一届2014年题目A题小鼠视觉感受区电位信号(LFP)与视觉刺激之间的关系研究B题机动目标的跟踪与反跟踪C题无线通信中的快时变信道建模D题人体营养健康角度的中国果蔬发展战略研究E题乘用车物流运输计划问题第十二届2015年题目A题水面舰艇编队防空和信息化战争评估模型B题数据的多流形结构分析C题移动通信中的无线信道“指纹”特征建模D题面向节能的单/多列车优化决策问题E题数控加工刀具运动的优化控制F题旅游路线规划问题第十三届2016年题目A题多无人机协同任务规划B题具有遗传性疾病和性状的遗传位点分析C题基于无线通信基站的室内三维定位问题D题军事行动避空侦察的时机和路线选择E题粮食最低收购价政策问题研究数据来源:。

宠物犬遗传性状基因分析犬种繁多,每个犬种都具有不同的外貌和性格特征。
这些特征是由遗传基因决定的。
对于宠物犬主人而言,了解犬只的遗传性状有助于选择适合自己的宠物,了解可能患病的风险以及培养正确的行为。
遗传性状是指在生物个体身上可以观察到的性状,如体毛颜色、眼睛颜色、耳朵形状等。
这些性状由基因在遗传过程中转移到后代的过程中决定。
基因是DNA分子中的一小段序列,它们通过蛋白质的合成来决定个体的特征。
对于宠物犬的基因分析,有几个主要方面需要考虑。
首先是外貌特征,比如体毛颜色、体型大小、耳朵形状等。
这些性状的遗传方式可能是显性遗传或隐性遗传。
显性遗传是指只需要从一个父母获得一个相关基因即可表现出来的性状,而隐性遗传是需要从两个父母都获得相关基因才能表现出来的性状。
例如,金毛犬的金黄色毛皮是由一个显性基因决定的。
只要一个金毛犬父母具有该基因,他们的后代就有可能拥有金黄色的毛皮。
然而,如果两个金毛犬父母都不是纯种金毛犬,那么他们的后代可能具有其他颜色的毛皮。
这是因为其他基因也可能影响一个性状的表现。
除了外貌特征,宠物犬的行为特征也受基因影响。
例如,一些犬种对主人忠诚,易训练,而其他犬种则更独立,不易训练。
这些行为特征也是由基因决定的。
然而,宠物犬的基因分析还可以用于检测潜在的健康问题。
某些犬种由于遗传缺陷,易患特定的疾病。
通过基因分析,可以确定宠物犬是否携带易患疾病的基因。
这样,主人可以提前采取预防措施或选择健康的犬只。
在进行宠物犬遗传性状基因分析时,常用的方法是基因测序。
基因测序可以确定宠物犬的DNA序列,进而确定其基因组中的特定基因。
此外,还可以使用PCR扩增等技术,快速检测特定基因的存在。
基因分析不仅有助于选择适合自己的宠物,了解可能患病的风险以及培养正确的行为,还对繁育纯种犬有着重要的意义。
繁育纯种犬需要保证其遗传特性的稳定性和一致性。
通过基因分析可以了解纯种犬的基因组信息,避免杂交繁育引入其他基因。

2016年全国研究生数学建模竞赛B题具有遗传性疾病和性状的遗传位点分析人体的每条染色体携带一个DNA分子,人的遗传密码由人体中的DNA携带。
DNA是由分别带有A,T,C,G四种碱基的脱氧核苷酸链接组成的双螺旋长链分子。
在这条双螺旋的长链中,共有约30亿个碱基对,而基因则是DNA长链中有遗传效应的一些片段。
在组成DNA 的数量浩瀚的碱基对(或对应的脱氧核苷酸)中,有一些特定位置的单个核苷酸经常发生变异引起DNA的多态性,我们称之为位点。
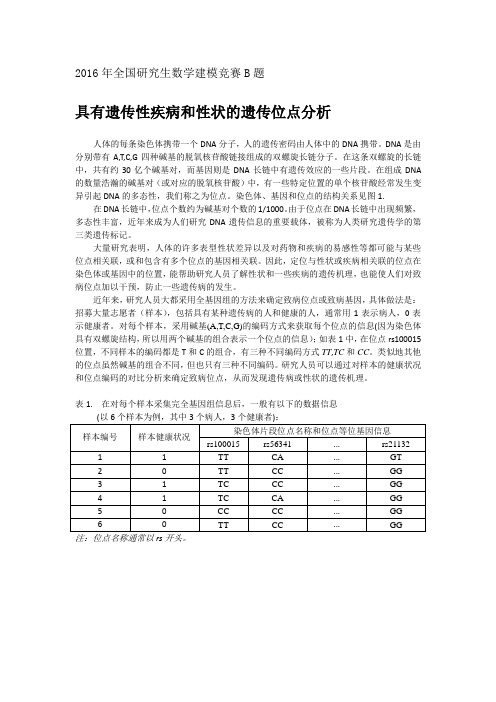
染色体、基因和位点的结构关系见图1.在DNA长链中,位点个数约为碱基对个数的1/1000。
由于位点在DNA长链中出现频繁,多态性丰富,近年来成为人们研究DNA遗传信息的重要载体,被称为人类研究遗传学的第三类遗传标记。
大量研究表明,人体的许多表型性状差异以及对药物和疾病的易感性等都可能与某些位点相关联,或和包含有多个位点的基因相关联。
因此,定位与性状或疾病相关联的位点在染色体或基因中的位置,能帮助研究人员了解性状和一些疾病的遗传机理,也能使人们对致病位点加以干预,防止一些遗传病的发生。
近年来,研究人员大都采用全基因组的方法来确定致病位点或致病基因,具体做法是:招募大量志愿者(样本),包括具有某种遗传病的人和健康的人,通常用1表示病人,0表示健康者。
对每个样本,采用碱基(A,T,C,G)的编码方式来获取每个位点的信息(因为染色体具有双螺旋结构,所以用两个碱基的组合表示一个位点的信息);如表1中,在位点rs100015位置,不同样本的编码都是T和C的组合,有三种不同编码方式TT,TC和CC。
类似地其他的位点虽然碱基的组合不同,但也只有三种不同编码。
研究人员可以通过对样本的健康状况和位点编码的对比分析来确定致病位点,从而发现遗传病或性状的遗传机理。
1表1. 在对每个样本采集完全基因组信息后,一般有以下的数据信息(以6个样本为例,其中3个病人,3个健康者):2基因位点染色体图1. 染色体、基因和位点的结构关系.本题目针对某种遗传疾病(简称疾病A)提供1000个样本的信息,这些信息包括这1000个样本的疾病信息、样本的9445个位点编码信息,以及包含这些位点的基因信息。

中国研究生数学建模竞赛试题汇总2021赛题汇总2021-A:相关矩阵组的低复杂度计算和存储建模2021-B:空气质量预报二次建模2021-C:帕金森病的脑深部电刺激治疗建模研究2021-D:抗乳腺癌候选药物的优化建模2021-E:信号干扰下的超宽带(UWB)精确定位问题2021-F:航空公司机组优化排班问题2020赛题汇总2020-A:芯片相噪算法2020-B:汽油辛烷值建模2020-C:面向康复工程的脑信号分析和判别建模2020-D:无人机集群协同对抗2020-E:能见度估计与预测2020-F:飞行器质心平衡供油策略优化2019赛题汇总2019-A: 无线智能传播模型2019-B:天文导航中的星图识别2019-C:视觉情报信息分析2019-D:汽车行驶工况构建2019-E:全球变暖?2019-F:多约束条件下智能飞行器航迹快速规划2018赛题汇总2018-A :关于跳台跳水体型系数设置的建模分析2018-B:光传送网建模与价值评估2018-C:对恐怖袭击事件记录数据的量化分析2018-D:基于卫星高度计海面高度异常资料获取潮汐调和常数方法及应用2018-E:多无人机对组网雷达的协同干扰2018-F:机场新增卫星厅对中转旅客影响的评估方法2017赛题汇总2017-A:无人机在抢险救灾中的优化运用2017-B:面向下一代光通信的VCSEL激光器仿真模型(华为命题)2017-C:航班恢复问题2017-D:基于监控视频的前景目标提取2017-E:多波次导弹发射中的规划问题2017-F:构建地下物流系统网络2016赛题汇总2016-A:多无人机协同任务规划2016-B:具有遗传性疾病和性状的遗传位点分析2016-C:基于无线通信基站的室内三维定位问题2016-D:军事行动避空侦察的时机和路线选择2016-E:粮食最低收购价政策问题研究2015赛题汇总2015-A:水面舰艇编队防空和信息化战争评估模型2015-B:数据的多流形结构分析2015-C:移动通信中的无线信道“指纹”特征建模2015-D:面向节能的单/多列车优化决策问题2015-E:数控加工刀具运动的优化控制2015-F:旅游路线规划问题2014赛题汇总2014-A:小鼠视觉感受区电位信号(LFP)与视觉刺激之间的关系研究2014-B:机动目标的跟踪与反跟踪2014-C:无线通信中的快时变信道建模2014-D:人体营养健康角度的中国果蔬发展战略研究2014-E:乘用车物流运输计划问题2013赛题汇总2013-A:变循环发动机部件法建模及优化2013-B:功率放大器非线性特性及预失真建模2013-C:微蜂窝环境中无线接收信号的特性分析2013-D:空气中PM2.5问题的研究2013-E:中等收入定位与人口度量模型研究2013-F:可持续的中国城乡居民养老保险体系的数学模型研究2012赛题汇总2012-A:基因识别问题及其算法实现2012-B:基于卫星无源探测的空间飞行器主动段轨道估计与误差分析2012-C:有杆抽油系统的数学建模及诊断2012-D:基于卫星云图的风矢场(云导风)度量模型与算法探讨2011赛题汇总2011-A:基于光的波粒二象性一种猜想的数学仿真2011-B:吸波材料与微波暗室问题的数学建模2011-C:小麦发育后期茎秆抗倒性的数学模型2011-D:房地产行业的数学建模2010赛题汇总2010-A:确定肿瘤的重要基因信息2010-B:与封堵溃口有关的重物落水后运动过程的数学建模2010-C:神经元的形态分类和识别2010-D:特殊工件磨削加工的数学建模2009赛题汇总2009-A:我国就业人数或城镇登记失业率的数学建模2009-B:枪弹头痕迹自动比对方法的研究2009-C:多传感器数据融合与航迹预测2009-D:110警车配置及巡逻方案2008赛题汇总2008-A:汶川地震中唐家山堰塞湖泄洪问题2008-B:城市道路交通信号实时控制问题2008-C:货运列车的编组调度问题2008-D:中央空调系统节能设计问题2007赛题汇总2007-A:建立食品卫生安全保障体系数学模型及改进模型的若干理论问题2007-B:机械臂运动路径设计问题2007-C:探讨提高高速公路路面质量的改进方案2007-D:邮政运输网络中的邮路规划和邮车调度2006赛题汇总2006-A:Ad Hoc网络中的区域划分和资源分配问题2006-B:确定高精度参数问题2006-C:维修线性流量阀时的内筒设计问题2006-D:学生面试问题2005赛题汇总2005-A:Highway Traveling time Estimate and Optimal Routing 2005-B:空中加油2005-C:城市交通管理中的出租车规划2005-D:仓库容量有限条件下的随机存贮管理2004赛题汇总2004A:发现黄球并定位2004B:实用下料问题2004C:售后服务数据的运用2004D:研究生录取问题。
2016年全国研究生数学建模竞赛B题具有遗传性疾病和性状的遗传位点分析人体的每条染色体携带一个DNA分子,人的遗传密码由人体中的DNA携带。
DNA是由分别带有A,T,C,G四种碱基的脱氧核苷酸链接组成的双螺旋长链分子。
在这条双螺旋的长链中,共有约30亿个碱基对,而基因则是DNA长链中有遗传效应的一些片段。
在组成DNA 的数量浩瀚的碱基对(或对应的脱氧核苷酸)中,有一些特定位置的单个核苷酸经常发生变异引起DNA的多态性,我们称之为位点。
染色体、基因和位点的结构关系见图1.在DNA长链中,位点个数约为碱基对个数的1/1000。
由于位点在DNA长链中出现频繁,多态性丰富,近年来成为人们研究DNA遗传信息的重要载体,被称为人类研究遗传学的第三类遗传标记。
大量研究表明,人体的许多表型性状差异以及对药物和疾病的易感性等都可能与某些位点相关联,或和包含有多个位点的基因相关联。
因此,定位与性状或疾病相关联的位点在染色体或基因中的位置,能帮助研究人员了解性状和一些疾病的遗传机理,也能使人们对致病位点加以干预,防止一些遗传病的发生。
近年来,研究人员大都采用全基因组的方法来确定致病位点或致病基因,具体做法是:招募大量志愿者(样本),包括具有某种遗传病的人和健康的人,通常用1表示病人,0表示健康者。
对每个样本,采用碱基(A,T,C,G)的编码方式来获取每个位点的信息(因为染色体具有双螺旋结构,所以用两个碱基的组合表示一个位点的信息);如表1中,在位点rs100015位置,不同样本的编码都是T和C的组合,有三种不同编码方式TT,TC和CC。
类似地其他的位点虽然碱基的组合不同,但也只有三种不同编码。
研究人员可以通过对样本的健康状况和位点编码的对比分析来确定致病位点,从而发现遗传病或性状的遗传机理。
表1. 在对每个样本采集完全基因组信息后,一般有以下的数据信息rs基因位点染色体图1. 染色体、基因和位点的结构关系.本题目针对某种遗传疾病(简称疾病A)提供1000个样本的信息,这些信息包括这1000个样本的疾病信息、样本的9445个位点编码信息,以及包含这些位点的基因信息。


实验组序号:日期:实验项目:人类性状的遗传分析实验目的:1.了解人类一些常见遗传性状的遗传方式。
2.了解群体控制不同遗传性状的基因分布情况。
实验原理:人类的遗传性状有许多是单基因性状,易于观察且具有典型的显隐性关系,在一定群体中进行调查,可以了解其遗传方式。
在自然界,无论动植物一种性别的任何一个个体有同样的机会与其相反性别的任何一个个体交配。
假设某一位点有一对等位基因A和a,A基因在群体出现的频率为p,a基因在群体出现的频率为q;基因型AA在群体出现的频率为D,基因型Aa在群体出现的频率为H,基因型aa在群体出现的频率为R。
群体(D,H,R)交配是完全随机的,那么这一群体基因频率和基因型频率的关系是:D=p2、H=2pq、R=q2。
根据Hardy-Weinberg定律由学生自行设计实验方案并加以实验。
实验对象:某群体实验用品: 1、器材玻璃棒、纱布块、乙醇棉球、镊子、量角器2、试剂 PTC1~14溶液、浓度递减实验内容与步骤1、卷舌在人群中,有的人能够卷舌,在近舌尖处两侧边缘向上甚至卷成管状,有的人则不能。
卷舌对不能卷舌为显性。
实验组成员间可互相观察,并记录下来,算出各类型所占的百分比,计算出该群体中相应的基因频率和基因型频率,记录观察结果,绘成系谱图,通过系谱分析其遗传方式。
2、耳垂形状人类耳垂可明显区分为有耳垂和无耳垂两种形状,前者为显性后者为隐形。
观察家庭成员的耳垂形状,看是否与上述遗传方式相符。
3、前额发际在人群中,有些人前额发际基本上属于平线,有些人在前额中部发际向下延伸呈峰形,试讨论这种形状属于哪种遗传方式。
4、将上述三个以及其他易于观察的形状的观察结果记录在附表上。
实验观察人类遗传性状与疾病关联调查表姓名曹雪班级麻醉112班学号6301611062。


内含子区域的遗传标记位点遗传标记位点是指在基因组中具有多态性的特定位点,可以用来标记遗传变异与特定性状之间的关联。
这些位点可以是单核苷酸多态性(SNP)、插入/缺失(InDel)或串联重复序列等。
内含子是基因组中的非编码DNA序列,其位于基因的外显子之间。
内含子通常被认为是无功能的,但近年来的研究发现,一些内含子与基因调控和疾病发生有关。
因此,研究人员开始关注内含子区域中的遗传标记位点,以探索其与疾病风险和表型变异之间的关系。
内含子区域的遗传标记位点在疾病遗传学和人类进化等研究领域具有重要作用。
首先,这些位点可以被用来进行关联分析,探究特定染色体区域与疾病之间的相关性。
例如,研究人员发现了一些内含子区域的遗传标记位点与癌症、心血管疾病和自身免疫性疾病等疾病的风险有关。
其次,内含子区域的遗传标记位点也可以用来推断人类的演化历史和族群间的遗传关系。
通过分析不同族群的内含子区域遗传标记位点的频率和多样性,可以重新构建人类不同种群的迁徙历史。
例如,研究人员利用内含子的遗传标记位点揭示了南北美洲原住民的迁徙历史以及不同族群之间的遗传关系。
此外,内含子区域的遗传标记位点也可以用于生物材料鉴定和个体识别。
由于内含子区域的遗传标记位点具有高度个体特异性和高变异性,可以通过分析这些位点的基因型信息来鉴定样本的来源。
这对于犯罪调查和人类遗骸鉴定等领域具有重要意义。
然而,内含子区域的遗传标记位点也存在一些技术和生物学上的挑战。
首先,内含子区域的遗传标记位点通常具有更高的多样性和复杂性,因此需要更高的测序深度和数据分析技术。
其次,内含子区域的功能和调控机制仍然不完全清楚,因此如何解释内含子区域的遗传标记位点与表型变异之间的关系仍然是一个挑战。
总的来说,内含子区域的遗传标记位点在疾病遗传学、人类进化和鉴定等领域具有重要的应用潜力。
随着高通量测序技术的发展和研究方法的完善,我们可以期待更多关于内含子区域的遗传标记位点的发现和功能解析,对于理解遗传变异与疾病发生机制以及人类进化的过程将有重要的启示作用。

人类性状的遗传分析一、实验目的1.了解人类一些常见遗传性状的遗传方式。
2.了解群体控制不同遗传性状的基因分布情况。
二、实验原理人类的遗传性状有许多是单基因性状,易于观察且具有典型的显隐性关系,在一定群体中进行调查,可以了解其遗传方式。
在自然界,无论动植物一种性别的任何一个个体有同样的机会与其相反性别的任何一个个体交配。
假设某一位点有一对等位基因A和a,A基因在群体出现的频率为p,a基因在群体出现的频率为q;基因型AA在群体出现的频率为D,基因型Aa在群体出现的频率为H,基因型aa在群体出现的频率为R。
群体(D,H,R)交配是完全随机的,那么这一群体基因频率和基因型频率的关系是:D=p2、H=2pq、R=q2。
三、实验方案通过查相关资料可知:人类常见遗传性状的识别-(表1)观察计划:我选择了10种容易看到的遗传性状作为观察内容。
⒈双眼皮还是单眼皮;⒉有无酒窝;⒊额头的头发是突出发际还是平发际;⒋舌头能否纵向卷成槽状(舌头向外伸出时);⒌有无耳垂;⒍食指与无名指长短的差别(谁长);⒎大拇指竖起时,能否后弯;8左右手哪个拇指在上(当两手相握时);9耳垢是干的还是湿的10血型步骤:⑴自我观察。
通过镜子先自我观察自身的上述10种遗传性状,将观察结果记入观察记录表。
⑵同学之间互相观察。
观察班上同学的上述10种遗传性状,将观察结果记入观察记录表。
⑶家庭成员观察。
观察自己的父母、祖父母、外祖父母的上述10种遗传性状,将观察结果记入观察记录表。
(表2)观察记录表四,结果分析:1.选择单双眼皮来分析,由表1可知双眼皮是显性,单眼皮是隐性。
由表2知aa=35.48% 开根号得a的频率为0.596,所以A=1-a=0.404,得AA=0.163, 2Aa=0.482, 所以aa+2Aa+AA=1正好验证了Hardy-Weinberg定律2.同样选取某同学家系单双眼皮性状做系谱分析祖父(双)——祖母(双)外祖父(双)——外祖母(双)││父(单)——————————————母(双)本人(单)由该家庭系谱图得知,由于本人和父亲都是单眼皮,基因型都应该是隐性纯合体d d;母亲的基因型肯定是杂合体D d。

SNP位点数据分析和人类遗传学研究SNP (Single Nucleotide Polymorphism) 位点数据分析和人类遗传学研究随着现代技术的快速发展,生物信息学领域的研究变得越来越重要。
其中,单核苷酸多态性(SNP)位点数据分析在人类遗传学研究中起着关键作用。
本文将讨论SNP位点的概念、分析方法以及其在人类遗传学研究中的应用。
首先,SNP位点是人类基因组中最常见的突变形式。
它是DNA序列中的单个核苷酸发生变异的地方,包括碱基的替换、插入和删除。
SNP位点通常在基因和表达调控区域中,对个体间的遗传差异和基因功能起着重要作用。
因此,研究SNP位点对于理解人类遗传学和疾病的发生机制至关重要。
在SNP位点数据的分析中,最常见的方法是基因型和等位基因频率分析。
基因型分析涉及确定每个个体的等位基因组合,包括纯合子(两个等位基因相同)和杂合子(两个等位基因不同)。
等位基因频率分析则是研究一个等位基因在某个群体中的频率。
通过这些分析方法,我们可以了解SNP位点的遗传多样性及其在人群间的分布情况。
此外,SNP位点数据还可以通过关联分析来研究基因与特定性状或疾病之间的联系。
关联分析(Association Analysis)是将SNP位点与某个性状或疾病之间的关联关系联系起来。
这种方法被广泛应用于复杂性疾病的研究,如肿瘤、心血管疾病和神经退行性疾病等。
通过关联分析,我们可以发现与某个特定性状或疾病相关的SNP位点,进一步了解其遗传机制,发现相关基因以及相关通路,为疾病的预测、诊断和治疗提供重要的线索。
SNP位点数据的分析离不开高通量测序技术的支持,如基因芯片和下一代测序。
这些技术的发展使得大规模SNP位点分析成为可能,相对应的数据处理和分析方法也在不断更新和改进。
然而,SNP位点数据分析中也存在一些挑战和限制,如缺乏样本数量和SNP位点的不均匀分布,这些问题需要继续研究和解决。
总结起来,SNP位点数据分析在人类遗传学研究中具有重要作用。

孟德尔遗传规律的应用(一)孟德尔遗传规律及其应用什么是孟德尔遗传规律?孟德尔遗传规律是指奥地利科学家格雷戈尔·约翰·孟德尔于1865年通过对豌豆的实验,发现了遗传物质的传递规律。
他观察了豌豆的性状,并通过对后代的分析,总结出了三个基本遗传规律,即“单一性状的分离规律”、“自由组合规律”和“分离组合规律”。
孟德尔遗传规律的应用1. 作物遗传改良通过对孟德尔遗传规律的研究,可以预测和改良作物的遗传性状。
农业科学家可以根据所需的特定性状,选择具有相应基因的亲本进行杂交,并根据单一性状的分离规律,通过观察后代的表现,筛选出符合要求的品种。
这种方法被广泛应用于作物品种改良,提高作物的产量、抗病性和品质。
2. 遗传疾病的研究孟德尔遗传规律的研究对于遗传疾病的认识和治疗也起到了重要作用。
通过了解某种疾病的遗传规律,可以预测个体是否会患病,从而进行早期干预和治疗。
同时,对于某些遗传疾病的病因研究,也可以通过对孟德尔遗传规律的应用,揭示该疾病的遗传机制,为研发新的治疗方法提供指导。
3. 基因工程基因工程领域广泛运用了孟德尔遗传规律。
科学家可以通过将外源基因引入目标生物体中,根据孟德尔的自由组合规律,通过遗传交叉和分析后代的性状,筛选出具有所需特性的基因组合,并进一步进行基因编辑和调控,实现对生物性状的精确改良。
4. 动物育种孟德尔遗传规律对于动物育种也起到了重要的指导作用。
通过对染色体的分离组合规律的研究,可以进行家畜的品种改良。
例如,在奶牛的育种中,根据孟德尔的分离组合规律,选取具有优良产奶性状的亲本进行杂交,筛选出高产奶的后代。
这种方法可以大大提高畜禽的经济效益和品质。
总结孟德尔遗传规律的发现和应用,为遗传学和生物学领域的研究和应用带来了革命性的进展。
通过深入研究和应用这些规律,不仅可以改良作物和提高动物品质,还可以加深对遗传疾病的认识和治疗,促进基因工程的发展。
孟德尔遗传规律的应用将持续推动生命科学领域的进步。

遗传类知识点总结1. 遗传物质遗传物质是决定生物遗传特征的物质,它主要存在于生物体的细胞核内,由DNA和RNA 组成。
DNA是脱氧核糖核酸,是细胞内遗传信息的主要载体,它通过遗传密码来控制生物体的形态、生长和生殖等生命活动。
RNA是核糖核酸,它主要参与蛋白质的合成过程,在生物体内具有重要的生物学功能。
2. 遗传信息的传递遗传信息的传递是指生物体内遗传物质的传递和传播过程,它主要通过细胞有丝分裂和生殖细胞的减数分裂来实现。
细胞有丝分裂是指细胞内的DNA复制和分裂过程,它保证了细胞遗传信息的完整传递。
生殖细胞的减数分裂是指生殖细胞在生殖器官内的分裂过程,它通过随机组合和配对的方式,使遗传物质在生殖细胞间的传递过程中产生了遗传变异,从而为物种的进化提供了遗传基础。
3. 基因和等位基因基因是决定生物性状的单元,它是遗传信息的基本单位。
等位基因是指同一基因在不同个体中的各种不同形态,它决定了生物体的遗传变异和多样性。
在自然界中,每个生物体的基因型都是由两个等位基因决定的,它们分别来自于父母亲的遗传。
4. 孟德尔的遗传规律孟德尔是遗传学的创始人,他通过豌豆杂交实验,发现了遗传领域中的一些基本规律,被称为孟德尔的遗传规律。
孟德尔的遗传规律包括基因的分离规律、基因的自由组合规律和基因的同源性规律,这些规律揭示了基因在遗传过程中的传递和表达规律,为后人对遗传学知识的深入研究提供了重要的理论基础。
5. 遗传性状的表现遗传性状的表现是指生物体内基因表达产生的生物学特征,它受到许多因素的影响,如基因型、环境、遗传背景等。
在生物体内,遗传性状主要通过基因的转录和翻译来表达,它决定了生物体的形态、生长和发育等生命活动。
6. 基因突变基因突变是指遗传物质中的基因发生了永久性的改变,这种改变可能导致生物体的遗传性状发生变异。
基因突变的产生可以通过物理、化学、生物等多种因素引起,它是生物体遗传变异和多样性的重要原因之一。
7. 遗传性疾病遗传性疾病是由基因突变引起的一类疾病,它在生物体内以遗传方式传递和表现。

基因位点描述一、什么是基因位点?基因位点是指基因组中特定位置的碱基序列,也可以理解为基因组中的一个“标记”。
位点的特点是具有多态性,即在不同个体中存在着不同的碱基组合。
基因位点和个体的基因型密切相关,对于疾病的发生、个体的性状表现等具有重要的影响作用。
二、基因位点的分类根据其在基因组中的位置和功能,基因位点可以分为以下几类:1. 基因座:基因座也称作基因位,是指位于染色体上的特定区域,包含着一个或多个基因序列。
基因座通常以字母和数字的组合来表示,如A1、B2等。
2. SNPs:SNPs,即单核苷酸多态性,是指在基因组中存在的单个碱基变异。
这种变异通常在不同个体中以等位基因的形式存在,可导致蛋白质结构或功能的变化。
3. SSRs:SSRs,即简单重复序列,是由短的核苷酸序列单元重复排列而成的序列。
SSRs通常在基因座的非编码区域中发现,具有高度的多态性。
4. CNVs:CNVs,即拷贝数变异,是指染色体上的片段重复或缺失导致基因拷贝数发生变化。
CNVs在人类个体中广泛存在,与多种疾病的发生有关。
三、基因位点的分析方法1. PCR:PCR,即聚合酶链式反应,是一种常用于从DNA样本中扩增特定位点的方法。
通过PCR扩增后,可以通过凝胶电泳等技术来检测位点是否存在及其多态性。
2. 测序:基因位点的测序是一种精确确定位点碱基序列的方法。
目前常用的测序技术包括Sanger测序和下一代测序技术,能够高效地获取大量位点信息。
3. 芯片技术:芯片技术是一种高通量的基因位点分析方法,通过将大量位点的探针固定在芯片上,可同时检测多个位点的多态性。
四、基因位点的应用1. 遗传病研究:基因位点的多态性与遗传病的发生密切相关。
研究人员可以通过分析基因位点与遗传病之间的关联性,揭示遗传病的发病机制,为疾病诊断和治疗提供依据。
2. 个体鉴定:基因位点的多态性可以用于个体鉴定。
通过检测个体的基因位点多态性,可以确定其独特的遗传特征,用于刑事侦查、亲子鉴定等领域。

第33卷第2期Vol.33No.2荆楚理工学院学报JournalofJingchuUniversityofTechnology2018年4月Apr.2018收稿日期:2018-01-02作者简介:邵缠婵(1993-)ꎬ女ꎬ安徽六安人ꎬ西北民族大学硕士研究生ꎮ研究方向:肿瘤学与数据可视化ꎮ基于统计方法的遗传性疾病基因位点分析邵缠婵1ꎬ余新林2(1.西北民族大学医学院ꎬ甘肃兰州㊀730000ꎻ2.甘肃省第二人民医院肿瘤介入科ꎬ甘肃兰州㊀730000)摘要:DNA的的碱基对中位点的改变经常会引发起各种各样的性状和疾病ꎮ文章利用R软件ꎬ运用统计学方法ꎬ参考各种优秀算法ꎬ建立相应模型ꎬ解决位点信息的编码问题㊁单个遗传疾病致病位点及多个遗传病关联位点的寻找问题㊁单个遗传疾病关联基因的寻找问题ꎬ并对实证结果进行可靠性检验ꎮ关键词:遗传性疾病ꎻ遗传统计ꎻ基因位点ꎻ因子分析中图分类号:R311㊀㊀文献标志码:A㊀㊀文章编号:1008-4657(2018)02-0047-060㊀引言随着人类基因组计划测序(TheHumanGenomeProjectꎬHGP)[1]的完成ꎬ人类基因组30亿碱基对的序列被公诸于世ꎮ在DNA链上一些特定位置的单个核苷酸具有多态性ꎬ经常发生变异[2]ꎬ从而引起DNA的多态性ꎮ据研究ꎬ在DNA长链中ꎬ位点个数约为碱基对个数的1/1000ꎬ即在整个人类基因组中ꎬ约有几百万个位点[3]ꎮ位点也因其在DNA长链中出现频繁性及自身的多态性ꎬ近年来成为人们研究DNA遗传信息的重要载体ꎮ大量研究表明ꎬ人体的许多表型性状差异以及对药物和疾病的易感性等都可能与某些位点相关联ꎬ或和包含有多个位点的基因相关联ꎬ例如冠心病[4]㊁糖尿病[5]等常见的疾病ꎮ因此ꎬ定位与疾病相关联的位点在基因中的位置ꎬ能帮助研究人员了解一些疾病的遗传机理ꎬ也能使人们对致病位点加以干预ꎬ防止一些遗传病的发生ꎮ近年来ꎬ研究人员大都采用全基因组的方法来确定致病位点或致病基因ꎬ具体做法是:招募大量志愿者(样本)ꎬ包括具有某种遗传病的人和健康的人ꎬ通常用1表示病人ꎬ0表示健康者ꎮ对每个样本ꎬ采用碱基(AꎬTꎬCꎬG)的编码方式来获取每个位点的信息(因为染色体具有双螺旋结构ꎬ所以用两个碱基的组合表示一个位点的信息)ꎮ通过对样本的健康状况和位点编码的对比分析来确定致病位点ꎬ从而发现遗传病的遗传机理ꎮ基于这一理论ꎬ本文借用2016年全国研究生数学建模竞赛B题的附件数据展开相关研究ꎬ尝试构建分析遗传性疾病遗传位点的方法体系ꎮ其中ꎬ附录数据包含某遗传疾病A的1000个样本的信息:样本的疾病信息(phenotype.txt)㊁样本的9445个位点编码信息(genotype.dat)㊁样本位点的基因信息(gene_info)㊁样本的10种相关疾病信息(multi_phenos.txt)ꎮ1㊀数据预处理在寻找具有遗传性疾病和性状的位点之前ꎬ鉴于原数据采用的碱基对编码形式难以进行数值㊁统计分析ꎬ本文采用编码转化的方法将样本的位点编码信息(genotype.dat)中每个位点的碱基(AꎬTꎬCꎬG)编码方式转化成数值编码方式ꎬ便于进行数据分析ꎮ具体的编码转化方法为:编码转化方式应该有利于数值计算ꎬ有利于之后问题的解决ꎬ考虑到每个位点只有3种形式的碱基组合ꎬ同时又要反映样本数据中患病组与未患病组在各个位点上的结构差异ꎮ本文考虑采取0ꎬ1ꎬ2的方式对每个位点分别进行编码ꎬ编码的规则如下表1所示ꎮ74表1㊀编码转化规则表序号位点含有的碱基编码转化规则序号位点含有的碱基编码转化规则1AꎬCAAң0ꎬACң1ꎬCCң24CꎬTCCң0ꎬCTң1ꎬTTң22AꎬTAAң0ꎬATң1ꎬTTң25CꎬGCCң0ꎬCGң1ꎬGGң23AꎬGAAң0ꎬAGң1ꎬGGң26TꎬGTTң0ꎬTGң1ꎬGGң22㊀实证分析㊀㊀为了寻找疾病或性状的致病位点ꎬ需要将患病组和未患病组的碱基进行对比并找出其差异性ꎮ目前学术界有很多优秀的方法可以反映患病和未患病组编码信息差异ꎬ例如蚁群算法[6]㊁基于智能算法的SNP选择方法[7]等ꎬ然而这些方法存在着耗时长㊁耗力大㊁程序运行时间过长㊁占用内存过大等问题ꎬ推广难度大ꎬ并且其得到的运算结果与一些简单的方法所得出的结果大致相同ꎮ因此ꎬ本文尝试建立一个简便易行且精确的模型体系ꎮ2.1㊀单个遗传病致病位点的寻找2.1.1㊀模型构建考虑极端的情况ꎬ假设在某一位点上ꎬ未患病组的碱基与患病组的碱基完全不一样ꎬ即未患病组可能是AA㊁ACꎬ而患病组可能全是CCꎬ此时认为该位点上未患病组和患病组的变异达到了最大ꎬ那么该位点便极有可能是致病位点ꎮ如果一个位点变异越大ꎬ那么该位点成为致病位点的可能性就越大ꎮ由于致病位点未患病组与患病组的编码信息存在较大差异ꎬ因此利用碱基出现的频数反映未患病组和患病组的差异也就成了可能ꎮ其计算方法如式(1):zi=ð10j=1yij-xij(1)其中ꎬxij(i=1ꎬ2ꎬ3ꎬ ꎬ9445ꎬj=1ꎬ2ꎬ3ꎬ ꎬ10)表示AAꎬCCꎬTTꎬGGꎬACꎬATꎬAGꎬCTꎬCGꎬTG在未患病组第i个位点上出现的的频数ꎻyij(i=1ꎬ2ꎬ3ꎬ ꎬ9445ꎬj=1ꎬ2ꎬ3ꎬ ꎬ10)表示AAꎬCCꎬTTꎬGGꎬACꎬATꎬAGꎬCTꎬCGꎬTG在患病组第i个位点上出现的的频数ꎻzi表示第i个位点的差异ꎮ比较式(1)中各位点的zi值大小ꎬ值越大表示该位点是致病位点的可能性越大ꎮ2.1.2㊀实证结果本文以genotype.dat(该数据包含500个未患某疾病的样本的9445个位点的碱基对信息和500个患上该疾病的样本的9445个位点的碱基对信息)的原始数据为例ꎬ对患病组和非患病组的样本分别统计各个位点上各碱基出现的频数ꎬ并使用R软件对式(1)进行计算得到实证结果ꎮ实证结果以散点图的形式呈现ꎬ如图1所示ꎬ其中zz表示各个位点的差异值ꎬindex表示位点的索引ꎬ即第几个位点ꎮ图1㊀单个遗传病致病位点的寻找结果图谱84根据图1ꎬ本文考虑将zz大于110的列为可能致病位点ꎬ从而找到了最有可能的位点索引序号为4938ꎬ通过位点名称与索引序号一一对应的关系ꎬ找到了4938的位点名称为rs2273298ꎮ同时ꎬ也找到了其他的可能致病点位ꎬ位点名称分别为rs880801ꎬrs4391636ꎬrs1541318ꎬrs7368252ꎬrs3013045ꎬrs4646092ꎬrs2143810ꎬrs1883567ꎬrs2807345ꎬrs932372ꎬrs7543405ꎬrs9659647ꎬrs12145450ꎮ2.1.3㊀结果检验在寻找到可能的致病位点之后ꎬ本文继续建立多元线性回归模型验证这些位点的可靠性ꎬ进一步验证式(1)的正确性ꎮ其计算方法如式(2):y=β0+ðNi=1βixi(2)将原始数据中phenotype(该数据为500个未患病样本与500个患病样本的0ꎬ1数据ꎬ0代表未患有该疾病ꎬ1代表患有该疾病)的数据作为因变量ꎬ将编码以后的第757ꎬ962ꎬ1195ꎬ1541ꎬ2938ꎬ3772ꎬ4526ꎬ4932ꎬ5937ꎬ6794ꎬ7737ꎬ8380ꎬ8435ꎬ9424位点信息作为自变量进行回归ꎬ多元回归结果如表2所示ꎮ表2㊀单个遗传病致病位点寻找结果的检验变量估计系数标准误t值变量估计系数标准误t值常数项0.39361∗∗∗0.093844.195z[ꎬ4932]-0.041490.02180-1.903z[ꎬ757]-0.06753∗0.02777-2.431z[ꎬ5937]-0.032480.02111-1.539z[ꎬ962]0.07638∗∗0.023663.228z[ꎬ6794]0.09035∗∗∗0.024913.627z[ꎬ1195]-0.044080.02292-1.923z[ꎬ7727]0.12703∗∗∗0.033933.744z[ꎬ1541]0.06053∗∗0.025152.406z[ꎬ8380]-0.012430.02570-0.576z[ꎬ2938]0.11442∗∗∗0.023024.971z[ꎬ8435]0.06768∗∗0.024022.817z[ꎬ3772]0.043130.024511.76z[ꎬ9424]0.05295∗0.021672.443z[ꎬ4526]-0.06597∗∗0.02318-2.846㊀㊀注:z[ꎬ757]代表第757个位点ꎬ其余类似ꎮ从表2可以看出ꎬ第2938ꎬ6794ꎬ7737位点的系数值较大ꎬ且t检验的统计值很小ꎬp值十分显著ꎻ而第1195ꎬ3772ꎬ4932ꎬ5937ꎬ8380位点的系数值较小ꎬp值不显著ꎮ且从模型的整体回归结果来看ꎬF检验的p值小于2.2e-16ꎬ说明模型整体上是显著的ꎬ即这些位点对是否致病存在显著的影响ꎮ综上所述ꎬ可以将第2938ꎬ6794ꎬ7737位点作为最有可能致病的位点ꎬ即位点rs2273298ꎬrs2807345ꎬrs932372ꎮ最后ꎬ考虑到位点之间可能存在的多重共线性问题ꎬ将第一步得到的其它位点列为候补致病位点ꎮ2.2㊀单个遗传病关联基因的寻找2.2.1㊀模型构建基因可以理解为若干个位点组成的集合ꎬ遗传疾病与基因的关联性可以由基因中包含的位点的全集或其子集合表现出来[7]ꎮ基于此ꎬ为了寻找单个遗传病最有可能的关联基因ꎬ本文尝试采取统计方法比较各个基因对疾病的影响大小ꎬ将影响大小量化ꎮ以单个遗传病致病位点的寻找思路为基础ꎬ我们利用患病组与未患病组各个位点的碱基对频数统计结果来构建位点的致病贡献率指标ꎬ从而寻找到单个遗传病的关联基因ꎮ其计算方法如式(3):gxj=zi/max(zi)ꎬ(i=j)(3)其中ꎬgxj表示第j个位点的致病贡献率ꎬmax(zi)表示zi的最大值ꎬzi越大第j个位点就越可能成为致病位点ꎬ对致病的贡献率就越大ꎮ本文将最有可能致病的位点的贡献率设置为1ꎬ其余位点致病贡献率依据式(3)的计算结果ꎮ在得到各位点对疾病的致病贡献率后ꎬ需要将原始数据中的基因数据㊁位点数据和致病贡献率指标一一对应ꎮ由于基因包含多个位点ꎬ所以求解基因对疾病的影响力就是求该基因所含位点的综合贡献率ꎮ而且ꎬ为了减少位点个数对基因致病率的影响ꎬ本文采用平均贡献率的方式来表达基因对疾病的影响力ꎬ其计算方法如式(4):gxvi=(ᵑj=lij=higxj)1/ni(4)94其中ꎬgxvi(i=1ꎬ2ꎬ3ꎬ...ꎬ300)表示第i个基因的平均致病贡献率ꎬni为第i个基因所包含的位点个数ꎬhi为第i个基因的起始位点ꎬli为第i个基因的终止位点ꎮ由于致病贡献率为相对指标ꎬ采用几何平均数来计算基因的平均致病贡献率最为合理ꎮ2.2.2㊀实证结果依据式(4)计算出数据文件gene_info(包含300个基因所包含的位点信息及位点名称)中各个基因所对应的平均致病贡献率ꎬ即的数值ꎮ实证结果以散点图的形式呈现ꎬ如图2所示ꎬ其中index表示基因的索引ꎬgxv表示平均致病贡献率ꎮ图2㊀单个遗传病关联基因的寻找结果图谱依据图2结果ꎬ将gxv大于0.25的基因作为可能致病的基因ꎬ得到第19ꎬ51ꎬ70ꎬ102ꎬ217ꎬ235ꎬ293个基因是最有可能的致病基因ꎮ另外ꎬ对比图1和图2可知ꎬ单个位点对某疾病的影响力差别较大ꎬ表现为图1的位点都主要集中于底部ꎬ处于顶部的位点很少ꎻ而一旦将位点集合起来纳入基因考虑时ꎬ单个基因对某疾病的影响力差别变小ꎬ表现为图2中各基因点主要集中在gsv=0.15附近ꎬ呈均匀分布ꎬ说明了基因内位点与位点之间存在着相互作用ꎮ2.3㊀多种遗传病关联位点的寻找2.3.1㊀模型构建寻找多种遗传病整体的关联位点同样也是热点问题ꎬ如果可以证明多种遗传病之间相互独立ꎬ就可以采取式(1)方法计算每种遗传病最有可能关联的位点[8]ꎮ而当各种遗传病间关联程度较高时ꎬ整合多种遗传病的信息便成了关键ꎮ本文通过计算原始数据multi_phenos.txt(包含10种遗传病的信息ꎬ每一列数据代表一种遗传病ꎬ其中数值1代表拥有该遗传病ꎬ数值0代表不拥有该遗传病ꎬ共有1000个样本)中10种遗传病的相关关系系数发现ꎬ10种遗传病存在高度相关ꎬ两两之间的相关系数均大于0.6ꎮ基于此ꎬ本文考虑采用因子分析的方法将10种遗传病指标整合为一个指标ꎬ将该情况简化为单个综合遗传病关联位点的寻找问题ꎮ利用R软件对multi_phenos.txt中十种遗传病进行因子分析处理ꎬ因子分析后主成分的贡献率如表3所示ꎮ表3㊀方差贡献率表成份初始特征值合计方差的%累积%提取平方和载入合计方差的%累积%17.42374.23274.2327.42374.23274.23220.4334.32978.56130.3213.20981.77040.3123.11784.888..................05㊀㊀当10种遗传病转化成一个主成分时ꎬ其累积方差贡献率为74.232%ꎬ损失了近26%的信息ꎮ在因子分析的基础上ꎬ采用因子综合得分代表十种遗传病整体的情况与genotype.dat的编码数据进行绑定ꎬ并对绑定的数据(即样本信息)按照计算所得的因子得分高低进行升序排列ꎬ某个样本因子得分越高ꎬ表示他拥有的遗传病越多ꎮ2.3.2㊀实证结果将1000个经过排序的样本按照1:1比例分为遗传病较少组和遗传病较多组ꎬ并利用碱基频数计算遗传病较少组和遗传病较多组的差异大小ꎬ方法同单个遗传病致病位点的寻找一致ꎮ实证结果见图3所示ꎬindex表示位点索引ꎬzz4表示差异值ꎮ图3㊀多个遗传病关联位点的寻找结果图谱根据图3的结果ꎬ本文将zz4值大于100的位点认定为与十种遗传病最有可能关联的位点ꎬ它们分别是第1050ꎬ1167ꎬ5338ꎬ6007ꎬ7181ꎬ8064位点ꎬ名称"rs351617"ꎬ"rss780983"ꎬ"rs4920299"ꎬ"rs11573253"ꎬ"rs3218121"ꎬ"rs4360511"ꎮ2.3.3㊀结果检验在寻找到最有可能的致病位点之后ꎬ本文继续建立多元线性回归模型验证这些位点的可靠性ꎮ由于要验证的是十种遗传病整体与图3选出的位点之间的统计相关性ꎬ需要将每种遗传病单独作为因变量进行回归ꎬ验证方法同单个遗传病致病位点的检验方法一致ꎮ如果某个位点在10个检验中都没有通过检验ꎬ就可以排除该位点的致病可能性ꎬ检验结果如表4所示ꎮ表4㊀多个遗传病致病位点寻找结果的检验z[ꎬ1050]z[ꎬ1167]z[ꎬ5338]z[ꎬ6077]z[ꎬ7181]z[ꎬ8064]整体F统计量性状10.00273∗∗0.487330.562740.154990.0049∗∗0.01873∗0.0004性状20.00769∗∗0.620450.697880.076670.00956∗∗0.0461∗0.0023性状30.00177∗∗0.746990.462990.095430.00064∗∗∗0.00168∗∗<0.00001性状4<0.00001∗∗∗0.964560.308480.961870.00378∗∗0.138270.00001性状50.00041∗∗∗0.551260.575380.656140.00078∗∗∗0.01147∗0.00003性状60.0024∗∗0.49190.84930.33770.0048∗∗0.0140∗0.00059性状70.00003∗∗∗0.955060.097670.295130.01139∗0.00094∗∗∗<0.00001性状80.00018∗∗∗0.289620.418450.256380.001831∗∗0.1726430.00008性状90.00112∗∗0.738490.612140.052460.00129∗∗0.00081∗∗∗<0.00001性状100.00128∗∗0.439820.911350.247840.00003∗∗∗0.053790.00002㊀㊀通过比较十个验证结果发现ꎬ在任意一个检验结果中ꎬ第1167ꎬ5338ꎬ6007位点都通不过t值检验ꎬ而15其余三个位点都能在大部分验证模型中通过检验ꎬ因此考虑剔除第1167ꎬ5338ꎬ6007位点ꎬ认为第1050ꎬ7181ꎬ8064位点与十种疾病整体最有可能关联ꎬ即位点"rs351617"ꎬ"rs3218121"ꎬ"rs4360511"ꎮ3㊀结论本文使用基本的统计学方法替代复杂的算法和机器学习方法ꎬ简便快速地实现了单个遗传病致病位点的寻找㊁单个遗传病关联基因的寻找以及多个遗传病关联位点的寻找ꎮ该统计方法在数据众多的情况下ꎬ可以大大节省电脑运行的内存和时间ꎮ4㊀讨论本研究方法还存在一定的系统误差:将不同位点的异位碱基对(即CA和AC)视为相等效果的碱基对ꎬ而事实上不同位点上的异位碱基对是不可视为相同的ꎻ本文构建的致病贡献率指标本身不是十分精确的指标ꎬ因此会对结果的精度造成影响ꎻ将十个性状的信息整合为一个综合整体指标时ꎬ损失了近25%的信息ꎬ因此对结果的精度造成了影响ꎮ另外ꎬ结果的精度还易受到位点间相互关系的影响ꎬ但随着学者们对基因㊁位点的研究的不断深入ꎬ基因位点的信息关系不断被揭露ꎬ该方法得到的结果的精度也会随之提高ꎮ参考文献:[1]ConsortiumGS.InitialSequencingandAnalysisoftheHumanGenome[J].Natureꎬ2001ꎬ409(6822):860-921. [2]TheInternationalSNPMapWorkingGroup.AmapofHumanGenomeSequenceVariationContaining1.42MillionSingleNucleotidePolymorphisms[J].Natureꎬ2001ꎬ409(6822):928-933.[3]G.A.ThorissonandL.D.Stein.TheSNPConsortiumWebsite:PastꎬPresentandFuture[J].NucleicAcidsResearchꎬ2003ꎬ31(1):124-127.[4]SamaniNJꎬErdmannJꎬHallASꎬetal.Genome-wideAssociationAnalysisofCoronaryArteryDisease[J].NEnglJMedꎬ2007ꎬ357(5):443-453.[5]HerbertAꎬGerryNPꎬMcQueenMBꎬetal.ACommonGeneticVariantisAassociatedwithAdultandChildhoodObesity[J].Scienceꎬ2006ꎬ312(5771):279-283.[6]刘丹.蚁群算法在基因与疾病关联性分析上的应用与实验研究[D].西安:西安电子科技大学ꎬ2013:29-44. [7]曾金萍.基于智能算法的信息SNP选择方法研究[D].长沙:湖南大学ꎬ2012:35-43.[8]赵发林ꎬ张涛ꎬ李康.基于遗传算法的随机森林模型在特征基因筛选中的应用[J].中国卫生统计ꎬ2016ꎬ33(04):559-562ꎬ566.[9]游伟.基于支持向量机的基因选择算法研究[D].长沙:湖南大学ꎬ2010:40-46.[责任编辑:许立群](上接第46页)[8]A.ChouchoulasꎬQ.Shen.RoughSet-AidedKeywordReductionforTextCategorization[J].AppliedArtificialIntelligenceꎬ2001ꎬ15(9):843-873.[9]苗夺谦ꎬ胡桂荣.知识约简的一种启发式算法[J].计算机研究与发展ꎬ1999ꎬ36(6):681-684.[10]YRHuꎬLXDingꎬDTXieꎬetal.TheSettingofParametersinanImprovedAntColonyOptimizationAlgorithmforFeatureSelection[J].JournalofComputationalInformationSystemsꎬ2012ꎬ8(19):8231-8238.[11]CSBaeꎬWCYehꎬYYChungꎬetal.FeatureSelectionwithIntelligentDynamicSwarmandRoughSet[J].ExpertSystemswithApplicationsꎬ2010ꎬ37(10):7026-7032.[12]UCIMachineLearningRepository[DB/OL].(2018-02-28)[2018-03-15].http://archive.ics.uci.edu/ml/data ̄sets.html.[13]Weka3:DataMiningSoftwareinJava[EB/OL].(2018-02-28)[2018-03-15].http://www.cs.aikato.ac.nz/ml/weka/index.html.[责任编辑:许立群]25。

遗传学揭示基因与性状之间的关联基因与性状之间的关联一直是遗传学研究的重要课题之一。
通过揭示基因与性状之间的关联,我们可以更深入地了解我们自身以及其他生物的遗传特征,并且为各种遗传疾病的预防、治疗提供重要的指导。
本文将介绍遗传学是如何揭示基因与性状之间的关联的。
遗传学是研究基因和遗传现象的科学学科。
在遗传学中,我们使用遗传学方法来确定基因与性状之间的关联。
遗传学方法包括孟德尔遗传学、连锁分析、关联分析等。
孟德尔遗传学是遗传学的基石。
格里戈尔·孟德尔通过对豌豆杂交实验的观察和分析,提出了基因的法则,即性状在后代中的传递是通过基因的方式进行的。
他发现,某些性状是通过一对基因来控制的,并且这些基因可以遗传给后代。
孟德尔的发现为遗传学奠定了基础,也揭示出基因与性状之间的关联。
连锁分析是另一种常用的遗传学方法,用于确定基因与性状之间的关联。
连锁分析通过观察基因在染色体上的相对位置来研究基因之间的遗传连锁关系。
基因在同一染色体上越靠近,它们之间的连锁关系就越强。
利用连锁分析,我们可以确定染色体特定区域上的基因,从而了解这些基因与特定性状之间的关联。
关联分析是一种通过分析基因与性状之间的相关性来确定基因与性状之间的关联的方法。
在关联分析中,科学家通常会收集一组个体的基因组数据和相应的性状数据。
然后,他们使用统计学方法来寻找基因与性状之间的相关性。
关联分析可以帮助我们确定特定基因变异与某种性状之间的关联,并确定一个人是否有遗传疾病的风险。
利用这些遗传学方法,科学家们已经成功地揭示了许多基因与性状之间的关联。
例如,他们发现特定基因突变可以导致一些遗传性疾病,如囊性纤维化和遗传性失明。
此外,他们还发现某些基因变异与个体的生理特征和行为特征之间存在关联,如眼睛颜色、身高、智力等。
这些研究不仅增加了我们对基因与性状之间关联的理解,也为预防、治疗遗传性疾病提供了重要的依据。
遗传学揭示基因与性状之间的关联对医学和农业领域具有重要的应用价值。

位点和基因的关系位点和基因是遗传学中重要的概念,它们之间有密切的关系。
位点是指染色体上的特定位置,基因则是位点上携带的遗传信息。
下面将从位点和基因的定义、特点以及它们之间的关系来探讨这个问题。
首先,位点指的是染色体上的特定位置,也可以理解为DNA序列上的一个标记点。
每个人的染色体上都有成千上万个位点。
这些位点可以用字母表示,比如“A”、“T”、“C”、“G”等。
位点的特点是在不同个体之间可以有差异,这种差异被称为单核苷酸多态性(SNP)。
SNP是指在同一个位点上可以有不同的碱基出现,比如在某个位点上,一个人的DNA序列为“A”,而另一个人的DNA序列为“T”。
而基因是位点上携带的遗传信息,是能够决定个体性状的分子遗传单位。
基因由多个位点组成,它们按照特定的顺序排列,形成一个功能完整的DNA片段。
基因可以分为DNA基因和RNA基因两类,DNA基因是编码蛋白质的,而RNA基因则编码非蛋白质的功能分子。
基因的特点是具有遗传性和可变性。
遗传性意味着基因可以从父母代传递给子代,子代会继承父母的基因。
可变性指的是基因可以发生变异,即某个位点上的碱基发生改变,进而改变基因所编码的蛋白质或分子的结构和功能。
位点和基因之间存在密切的关系。
一方面,基因是位点上的遗传信息,位点的变异会导致基因的变异。
基因的变异可能影响蛋白质的结构和功能,进而影响个体的性状和表型。
例如,某个位点上的碱基发生变异,导致一个基因编码的蛋白质结构发生改变,可能会引起某种疾病的发生。
另一方面,通过对位点的检测和分析,可以揭示基因的组成和变异情况。
目前,科学家们已经可以通过高通量测序技术对个体的基因组进行快速测序,并通过对位点的分析来了解基因的变异情况。
位点和基因的关系对于遗传学的研究和应用具有重要的意义。
通过对位点和基因的研究,可以揭示基因与疾病之间的关系,帮助我们了解疾病的发生机制。
同时,位点和基因的研究还可以为个体的健康管理提供指导。
例如,通过对位点的检测和分析,可以预测个体可能患某种疾病的风险,进而采取相应的预防措施。

定量遗传学研究方法定量遗传学是一门研究遗传变异对性状变异影响的学科。
通过定量遗传学研究方法,我们可以了解物种中的遗传基础和性状表现的规律。
在这篇文章中,我将向大家介绍几种常用的定量遗传学研究方法。
1. 全基因组关联研究(GWAS)全基因组关联研究是一种用于发现遗传变异和性状关联的方法。
GWAS通过对大规模样本群体的基因组信息进行分析,可以找到与性状相关的遗传变异。
这种方法的优点是能够检测到较为小的遗传效应,但也存在一些局限性,比如可能会由于样本限制而忽略掉某些重要的变异。
2. 聚合分析聚合分析通过研究具有共同精神疾病或遗传异常的家族成员,来探索遗传因素与性状之间的关联。
这种方法适用于研究遗传基础不明确的复杂性状。
通过比较不同家族成员之间的遗传相似性,可以推断与性状相关的基因。
3. 在后代中测量遗传变异在后代中测量遗传变异是一种常用的研究方法,可以通过测量性状在不同后代中的变异程度来推断遗传效应。
通过对大量后代的观察和测量,可以估计不同基因型对性状变异的贡献程度。
4. 模拟和定量基因组学模拟和定量基因组学是一种通过计算模型来预测基因型与表现型之间关系的方法。
通过分析基因组中的遗传变异,结合统计学模型,可以预测不同基因型对性状的影响。
这种方法能够帮助我们了解不同基因型的遗传效应,并预测个体性状的可能范围。
5. 等位基因特异性表达分析等位基因特异性表达分析是一种研究变异基因型如何影响基因表达的方法。
通过分析不同基因型在基因表达水平上的差异,可以发现与性状相关的表达差异。
这种方法可以提供关于与性状有关的基因表达的信息。
以上是几种常见的定量遗传学研究方法。
这些方法提供了我们研究遗传变异对性状变异影响的有效工具。
通过深入研究遗传基础和性状表现的规律,我们可以更好地理解物种的遗传特性,并为遗传学疾病的研究和对性状的遗传改良提供理论基础。
这些研究方法的进一步发展,将为我们揭示性状多样性形成和进化的遗传机制提供更多的见解。

遗传标记分析的原理与应用随着科技的快速发展,遗传标记分析技术在生物学和医学领域中得到广泛应用,成为了许多研究工作的重要组成部分。
遗传标记分析可以从分子水平上研究基因的遗传规律和变异情况,有助于我们更好地了解人类、动植物群体的遗传信息,进而对我们的生命、环境、健康等诸多方面进行更准确和全面的探究。
一、遗传标记分析的原理遗传标记分析是将已知的特定遗传位点信息转化为可以利用的测量数据,以便检测、挖掘和分析这些位点的遗传变异情况。
遗传标记分析的实现基础是遗传多态性,包括基因多态性、染色体多态性、DNA序列多态性等。
在遗传标记分析中,最常用的两个遗传标记是基因多态性标记和分类标记。
基因多态性标记指的是利用多态性基因位点上的遗传变异来生成遗传标记。
这些位点通常是DNA序列中的与功能无关的重复序列(SSR)或单核苷酸多态性(SNP),之所以不能是具有功能的基因位点,是因为它们的变异不太可能对生物体的生理功能产生直接影响。
例如,考虑到人类DNA的大部分都是功能未知的非编码DNA序列,所以研究人类基因组的标记通常是SSR。
SSR是一种含有较短的(通常是1-6个核苷酸长度)DNA序列的重复序列,通过评估一个给定位点上的重复序列数量的变异,可以评估不同基因型之间的遗传差异。
分类标记是利用一个或多个定量性状或性状指标来生成的标记。
分类标记通常是定量性状的离散化,例如将身高分类为高、中、低三类,然后将这种分类信息作为标记将样本进行归类。
二、遗传标记分析的应用1.种群遗传学遗传标记分析可以用于研究基因在个体和群体水平的遗传变异,进而推断种群分化的历史、迁移和演化等问题。
例如,利用SSR标记的数据,可以研究鱼类种群的结构和分布,评估其生态重要性和保护策略,并进一步研究不同种群之间的关系和来源。
2.遗传疾病诊治遗传疾病是由基因突变引起的疾病,遗传标记分析可以用于识别导致遗传疾病的潜在基因。
通过检测大量患者和健康人群的基因型和序列数据,就可以发现在某些疾病中高频率的基因变异。

基因位点解读
基因是携带遗传信息的基本单位,其特定的一段DNA序列称为基因位点。
基
因位点的不同形式可能影响个体的特征和性状,因此对基因位点进行解读十分重要。
基因位点的定义
基因位点是基因组中的特定位置,通常由一系列碱基组成。
每个基因位点可能
存在不同的等位基因,这些等位基因会在个体中表现出不同的性状。
基因位点的重要性
1.遗传相关性:基因位点决定了个体的遗传信息,其突变可能导致遗
传疾病或其他影响。
2.个体差异:不同基因位点的组合可能导致个体之间的生理和行为上
的差异。
3.生物演化:基因位点的变异是种群进化和适应环境的基础。
基因位点的解读方法
1.测序技术:通过测序技术可以确定特定基因位点的碱基序列,进而
分析其潜在功能。
2.关联分析:研究人员可以通过分析大规模基因位点和表型数据的关
联,发现基因位点与特定性状之间的联系。
3.功能预测:利用生物信息学方法,可以推断基因位点可能的功能,
从而推断其对个体性状的影响。
基因位点在疾病研究中的应用
1.疾病风险评估:基因位点的变异与某些疾病的发生风险相关,通过
分析基因位点可以进行疾病风险评估。
2.药物反应预测:个体基因位点的不同可能会影响对特定药物的反应,
基因位点解读可以指导个体化用药。
结语
基因位点的解读是基因组学研究中的关键环节,对了解个体遗传信息、疾病发
生机制等具有重要意义。
随着技术的不断进步,我们对基因位点的解读和应用将变得更加深入和精准。
参赛密码(由组委会填写)“华为杯”第十三届全国研究生数学建模竞赛学校江苏科技大学参赛队号队员姓名1. 孙佳伟2. 李袁3. 李肇基参赛密码(由组委会填写)“华为杯”第十三届全国研究生数学建模竞赛题目具有遗传性疾病和性状的遗传位点分析摘要:本文设计了基于属性重要度的选择算法,并通过SVM分类器构建出预测模型,对不同的位点和基因进行分析,判断每个位点或基因对某种疾病的预测精度,从而判断是否为致病位点或者致病基因。
最后利用该算法和模型,预测出十种性状相关的致病位点。
问题1,针对每个位点有碱基对组成的性质,为了方面描述和分析,本文采用了十进制编码,每个位点的属性值可以通过0-9中的一个数进行表示,具体的编码格式,文中给出了详细的编码表。
问题 2,设计了基于属性重要度的特征选择算法,通过SVM分类器构建出预测模型,通过问题1中的特征表示方式,提取所有样本每列的特征,并对每列的特征属性进行重要度分析,进而判断该疾病与位点rs, rs,rs2486182,rs2274119,rs2235537相关。
问题 3,每个基因是由不同位点组成的集合,则每个基因的所有特征属性即对应集合里位点特征属性的集合,利用问题2优化的模型,通过预测精度,对每个基因的对某疾病的重要度进一步分析得出,该疾病与致病基因gene_171相关。
问题 4,利用本文提出的模型,对10种性状中的每种性状中,继续通过属性重要度分析,识别出在不同性状中最有可能的致病位点,最后得出10个形状的相关致病位点分别为:rs, rs935075, rs2840758, rs1855786, rs2647168, rs, rs744834, rs4920522, rs, rs。
本文亮点是,提出基于属性重要度的选择算法,通过SVM构建出预测模型,利用网格搜索进行寻优,判断每列属性的重要度,从而判断致病位点或者致病基因。
关键词:属性重要度;SVM分类器;优化模型;位点(SNPs)一问题重述问题1,请用适当的方法,把中每个位点的碱基(A,T,C,G)编码方式转化成数值编码方式,便于进行数据分析。
问题2,根据附录中1000个样本在某条有可能致病的染色体片段上的9445个位点[1]的编码信息(见和样本患有遗传疾病A的信息(见文件)。
设计或采用一个方法,找出某种疾病最有可能的一个或几个致病位点,并给出相关的理论依据。
问题3,同上题中的样本患有遗传疾病A的信息(文件)。
现有300个基因,每个基因所包含的位点名称见文件夹gene_info中的300个dat文件,每个dat文件列出了对应基因所包含的位点(位点信息见文件。
由于可以把基因理解为若干个位点组成的集合,遗传疾病与基因的关联性可以由基因中包含的位点的全集或其子集合表现出来请找出与疾病最有可能相关的一个或几个基因,并说明理由。
问题4,在问题二中,已知9445个位点,其编码信息见文件。
在实际的研究中,科研人员往往把相关的性状或疾病看成一个整体,然后来探寻与它们相关的位点或基因。
试根据文件给出的1000个样本的10个相关联性状的信息及其9445个位点的编码信息(见,找出与中10个性状有关联的位点。
二问题分析问题1的分析问题1提供的1000个样本,每个样本均有9445个位点,每个位点是一个碱基对,本文通过十进制对所有碱基对[2]进行编码,利用此特征表示方法,每个样本得到9445个属性,每个属性通过0-9中的数值进行表示。
问题2的分析问题2和问题3都是要利用某种方法,进行致病位点和致病基因的检测,预测某种疾病的致病位点。
预测该疾病相关的致病位点,其实就是判断不同位点对该疾病的影响程度,即判别每个位点的属性重要度。
本文设计了基于属性重要度的选择算法,并利用SVM构建预测器,计算每个位点对疾病的预测精度,从而得到与该疾病相关的致病位点。
问题3的分析问题3与问题2比较,问题2是识别与某疾病相关的致病位点,而某个基因是由多个位点组成的集合,问题3是识别某疾病相关的致病基因,为了识别致病基因,由于基因是由多个位点组成的集合,则基因的特征属性即多个位点特征属性组成的集合。
利用每个基因的特征属性,通过问题2中设计的算法和构建的预测模型,从而识别与某疾病相关的致病基因。
问题4的分析问题4中,人的某些疾病是和性状相关的,材料中提供了10种性状,要求判断与这10种性状的相关的致病位点。
可以对每个性状分析,通过问题2种设计的选择算法和构建的预测模型,识别出某个性状相关的致病位点。
三模型假设和符号说明模型假设(1)假设给出的样本数据能分别代表整个的正常和患病群体。
(2)忽略寻优时SVM分类器本身造成的偏差。
(3)给出的位点和基因均是有效的位点和基因。
符号说明(未说明)(1) A,C,G,T :DNA中的四种碱基(2) Acc :即accuracy,预测精度(3) PC:第n个位点的属性n(4) S :特征属性子集(5))f:通过十进制编码得到位点子集的特征属性(S(6):第i个位点的属性,在第1组属性选择中进行融合(7) Sig :即significant,属性重要度四、模型建立与求解问题1:每个位点碱基编码方式转化数据分析本文所使用位点测试数据集,来自1000个可能致病的染色体片段试验检测结果,标签分布为500个无病染色体使用0表示,500个患病染色体使用1表示,且每个致病染色体上有9445个碱基对,以此作为位点。
十进制编码本文采用十进制{0,1,2,...9}编码将每个碱基对转化成数据编码方式,以便于数据分析。
“AA”为“0”;“AC”为“1”;“AG”为2;“AT”为3...“TT”为9,详见碱基对编码表1(其中{AC,CA};{CG,GC};...碱基对表示方式相同)。
另外,位点中出现字符‘I’和‘D’,根据说明,分别用‘T’和‘C’代替问题2:找出与疾病最有可能相关的一个或几个位点位点属性矩阵由于所有样本序列上的本一个二核苷酸位点代表了一个属性,本文总共有9445个位点即9445个不同的属性,这些属性由十进制表示(见附图1)。
其中,属性列中PC1~PCn表示9445个不同的属性指标;AA,AC,AG,AT,...,TT表示16中不同的原始二核苷酸。
..................9 1 ... 89 4 ... 76 4 ... 76 1 ... 74 4 ... 79 4 (7)rs10015rs5641rs21132pc 1pc 2pc n图1 十进制编码编码碱基对实验测试方法和分类器设计1. 实验测试方法K 折交叉验证、Jackknife (留一法)测试和独立数据集测试是三种常用的实验测试方法。
在本试验中采取了K 折交叉验证,K 折交叉验证是指经过初始采样后将初始样本分割成K 个子样本,然后选择一个单独的子样本作为测试数据,剩下的K-1个样本用来训练模型。
将这个交叉验证重复K 次,使得每个子样本都验证一次,将K 次结果取平均,得到一个估测结果。
这个方法优势在于随机产生的子样本的训练和验证能够同时进行,每次的结果验证一次,本文采用了5折交叉验证。
2. 分类器设计特征向量提取完成后便要面临对向量的分类问题,选取一个合适的分类器,将大大提高相关识别问题的精度,然而分类器研究至今出现了许多,例如贝叶斯分类器[3],支持向量机(SVM )[4,5,6,7],K 近邻分类器[8,9,10]以及DeepLearning [11,12]。
在本篇论文中,我们采用支持向量机对样本进行分类。
支持向量机是由Vapnik 等根据统计学习理论提出的,在考虑结构风险最小化原则和VC 维理论的基础上,在有限的样本信息中,寻找模型复杂性和学习能力的最佳折中点。
这个分类方法在解决模式分类与非线性映射问题中非常适用。
例如图2便是在二维平面中一个分类,线AB 上的点组成的向量就是这个样本的支持向量,然而在多数情况下数据样本将不仅仅局限于二维,通常都是在高维空间中的样本,如图3,这时的支持向量是以面的形式表现出来。
图2 支持向量机二维原理图图3 支持向量机高维原理图在生物信息学中,非线性以及高维数据的处理十分常见,所以支持向量机在生物信息学的领域被广泛使用。
因此本篇文章中,包作为支持向量机的实现。
由于在非线性分类过程的有效性和速度,因为径向基核函数(RBF)利用网格搜索法优化了正则化参数和核参数,所以我们使用这个函数对样本进行预测。
从支持向量机获得的概率得分被用来预测本次实验的最终结果。
评价标准为了对我们的预测方法进行合理的评价,我们在考虑结果的准确性上,加入了相关系数,用这些数据对预测结果进行一系列评价。
本文使用下列度量模型的性能进行了评价:准确度(Acc),它可以表示为(1)其中,TP 表示被正确判断为正样本的正样本数;FP 表示被错误判断为正样本的负样本数;TN 表示被正确判断为负样本的负样本数;FN 表示被错误判断为负样本的正样本数。
将公式(1)化简,并定义新的字符表示,化为如下形式:(2)用N ++代替TP ,N +-代替FN ,N --代替TN ,N -+代替FP ,显然N N N ++++-=+,N +表示所有的正样本数。
同理N N N ----+=+,N -表示所有的负样本数。
根据此式,我们可以很容易的得出以下的结构。
当N +-=N -+=0时,即所有的样本都被正确判断时,Acc 1=;当N N ++-=且N N --+=时,即所有的样本都被错误判断时,Acc 0=;Acc 是对整个数据集的评价指标。
所以我们在比较预测器性能的时候,主要对比Acc 的值。
基于属性重要度的选择算法设计1. 属性重要度问题1中通过十进制编码方法,对样本中9445个位点进行特征表示,得到9445个特征属性,且通过实验发现,多个属性的组合来进行该模型的预测可能比单个属性预测的结果要更高,但是并不是所有的属性都与该疾病有必然的联系,可能存在冗余的属性,即可能存在冗余的位点,本文设计一种基于属性重要的选择算法,通过属性选择的方式可以有效的去除冗余属性,提高判断的准确性,从而保障了预测的可靠性。
2. 属性选择算法假设第n 列的属性记为PC n ,则全部9445个位点的属性集合为{PC 1,PC 2...PC n …PC 9445},选取位点属性子集为S ,通过问题1中的编码方式编码样本序列特征记为)(S f 。
))S ((Acc }))PC {S ((Acc )PC ,S (Sig n n f f -+= (3)公式(3)中的))((Acc S f 和}))PC {((Acc n S +f 分别表示,采用位点属性子集S 和性选择算法得到的子集}PC {n S +,利用SVM 构建的预测器的预测精度。
判断标准:(1)当Sig(S,PC n )>0,表明增加PC n 这列属性对模型预测性能的提高有积极的作用,其可以融合为多重属性并利用预测器进行下一步判断。