Highly Available Long Running Transactions and Activities for J2EE Applications
- 格式:pdf
- 大小:1.24 MB
- 文档页数:10

Ensign Tuning GuideQuantum Sails has used our years of experience building and racing Ensign sails to develop a fast set of Class sails, geared for performance in all racing conditions. Together with the industry’s most rigorous quality standards of Cloth Selection, Cutting Accuracy, and Craftsmanship, we have created a unique combination of speed, quality, and long lasting performance sails.We hope this guide will help you take your Ensign campaign all the way to the Grand Prix level. We believe that a successful racing campaign is a combination of many elements. While one of the most dramatic improvements to any Ensign is a new suit of Quantum sails, we cannot over emphasize the importance of raising the level of all the other components of your campaign to that of your new Quantum sails.Before Your Boat Hits The WaterClean and sand your bottom to 600 finish, flattening any bumps. It is important to make sure to keep your bilge as dry as possible at all times, especially before the boat is put away for the offseason. There is a fiberglass shell surrounding the Lead Ballast of your keel. Water will seep into this area and contract and expand in the heat or freezing cold respectively, cracking the shell. Cracks in the outer shell are slow, creating drag. Large cracks are potentially dangerous and should be brought to the attention of a professional boat shop.The other major reason for meticulous water concern is that the boats are constructed with a large amount of Balsa wood. Although lightweight, Balsa absorbs water quickly like a sponge and will aid in rot, especially in the deck and cabinhouse. The RudderThe Ensign Rules state the Rudder Specifications thoroughly. Before glassing the rudder please consult your rule book. We prefer the fiberglass rudders over wood ones for better performance. Fiberglass is easier to fair to the rules and will not absorb water as easily.RiggingWe recommend your set up your headstay close to the Ensign Class maximum length (26’ 3 3/4”). The best All-Purpose length is 26’3”. This allows you to set up the rig with the proper rake and prebend required for Quantum Ensign sails. Refer to our Trim/Tuning Chart for specifics on the shroud tensions. (below)Mast ButtWhere the mast butt is located is at the heart of rig tuning. Because mast step locations vary from boat to boat, we do not have a concrete measurement of where your own butt should be located. What you want to look for is 1 ½ - 2” of prebend in the mast when the uppers and forward lower shrouds are tensioned properly (~40/40 units) with the rig centered and the backstay hooked up and reading 0-5 units of measure on the tuning gauge (using the gauge on the forestay). We usually recommend shimming your mast forward at the deck, if there is room to do so. If you have less than 2” of prebend, un-screw the shrouds and move the butt aft a little, or forward a little if the mast has too much bend. Re-tension the rig. This is an important step and if you are not confident with the pre-bend amount repeat these steps until you see this 2” of pre-bend. I usually just sight up the mast when looking for this.See Chart Included to help locate your Mast Butt in the proper position (last page).Upwind SailingGet the crew dialed into the trim/tuning chart. By changing gears in the varying conditions, big gains can be made. The sails are very versatile. In the lulls, move trim to the lower breeze settings in the main and genoa. In the puffs, trim the genoa and main for speed. If over-powered, ease the traveler down to flatten the boat and relieve weather helm.The Quantum Mainsail gives lots of kick for pointing. By bringing the top batten to parallel and then to windward a few degrees, the boat will point 3-6︒ higher. When the boat slows in speed, change from “point” back to “speed” mode by easing the mainsheet and bringing the traveler up, keeping the boom in centerline of the boat. This versatility in the main is very desirable for squeezing off competition behind you and not getting sucked in to competition in front of you.The mainsail needs the sheet to be eased hard to fall off at the weather mark and during “Ducking” other boats on the beat.Set up the genoa leads for the prevailing breeze and then play the sheet through the puffs and lulls. Have the crew sit forward and to leeward in light air. Then move them to the benches as the boat speeds up around 7 knots of wind and more. In lighter air the Genoa will be about 3” off the spreader and just touching the shr ouds at the foot. As the wind build the foot should be pulled in tighter on the foot and still about 2 - 3” off the spreader. You will want to move the genoa lead car aft as the wind builds and this will aid in twisting the genoa and de-powering.Downwind SailingTry to keep the top batten parallel to the boom by adjusting the vang. In reaching under genoa, allow the main to twist similar to the genoas Leech. Trim the Pole just aft of square to the apparent wind in light/medium and medium to heavy breezes. In light air square the pole to 90︒, in drifters try the pole just forward of square.The spinnaker should be flown with the clews relatively even to having the guy slightly higher by 2 - 3”. The chute performs best with a 4 - 6” curl in the wi ndward leech shoulder. When reaching hard try pulling the pole down more, this will pull the draft of the chute forward and aid in turning the symmetric spinnaker into more of an asymmetric.Heel the boat to weather when running dead downwind in a moderate breeze. In lighter wind heel the boat according to the helm feel – usually flat to slight leeward heel.Try to steer the boat with the crew weight instead of the rudder downwind. You want to minimize the water flow disturbance past the rudder, minimizing drag. Move the crew to weather to go down or fall off, and move the crew to leeward to come up. Crew weight is most effective moving around in the middle and forward of the companionway.OverallGood Luck and feel free to contact us with any questions and input. We realize that the boats can be setup quite differently, so we have endeavored to provide simple, general numbers for you to shoot for. Have a great season.Doug Burtner Randy Shore Allen Terhune Terry Flynn585-342-5200 401-849-7700 410-268-1161 281-474-4168 dburtner@... rshore@... aterhune@... tflynn@...…@ENSIGN Trim & Tuning Chart***NOTE- The stays are 5/32” diameter, and we use “Loos Gauge” 91-model A (not B)。


高加速寿命试验(Highly Accelerated Life Test-HALT/HASA/HASS) 加速应力试验源由(Accelerated Stress Test:简称AST/ALT),源起于1960年代美国因应太空计划对高可靠度的需求而被发展出来。
随着科技高度发展及快速变化的市场需求,过去耗时的产品验证方式已逐渐无法应付如此快速变化的市场需求进而影响到产品于市场之竞争力,因此,如何快速且有效发现产品设计缺陷并于设计阶段加以修正为现今国内外各大厂之主要关键问题,亦即是HALT&HASS逐渐被重视的原因。
众所皆知,产品在设计阶段进行缺陷修正是极为容易的,在大量生产后进行缺陷修正则困难度相对提高。
微利时代若产品在市场于保固期内出现缺陷则所花费成本与商誉损失将无法计算。
因此1990年代后以美国为首的国际各大厂(包括hp、Dell、Cisco、Nortel、Tetronix、Motorola等)均相继以HALT手法作为新产品开发阶段迅速找出产品设计及制造的缺陷同时改善缺陷已达降低保固期成本、增加产品可靠度并缩短产品上市时间。
同时可利用HALT所发现之失效模式与相关资做为后续研发产品的重要依据。
目前有航空电子、汽车及信息等高科技产业皆已投入HALT 领域之测试,并且已有相当成效。
基本概念:1)HALT :Highly AcceleratedLife Testing(高加速寿命测试)2)HASS:Highly AcceleratedStress Screen(高加速应力筛选)3)HASA:Highly AcceleratedStress Audit(高加速应力稽核)关于HALT:1)测试目的:在设计初期对已有的原型机或者工程样机进行应力测试并至失效,对于发现的失效进行分析改善,例如更换部件等方法,使得产品的设计更加强健,可靠性更高。
简单来说就是:测试->失效->分析->改善->测试....的循环,网上找了下面一张图以便于理解。

The I-Shift gearbox’s functions are optimised with specially adapted softwar e packages that make the gear box even more pr actical and economical by adapting the gearshift strategies to the current transport conditions.Sales variantsBasic version (TP-BAS)TP-BAS is the standard software package supplied with the I-Shift and includes the gearbox’s basic functions for allround driving.I-Shift distribution gear changing software (TP-DIST)TP-DIST adapts the gearbox’s function to the specific con-ditions in the distribution segment. The software package includes functions that aid manoeuvrability when starting off from standstill, in manoeuvring and when driving at low speed.I-Shift construction gear changing software (TP-CON)TP-CON adapts the gearbox’s function to the specific conditions in the construction segment. The software package includes functions that aid manoeuvrability when starting off from standstill, in manoeuvring and when driving at low speed. This software can also handle tougher road conditions.I-Shift long haul gear changing software (TP-LONG)TP-LONG includes intelligent functions that minimise fuel consumption. This software package is ideal for long-haul operations where strong emphasis is placed on fuel economy. This package includes the I-Roll function.Heavy duty transport (TP-HD)TP-HD optimises I-Shift for heavy duty transport with high gross combination weights (>85 tonnes). Regardless of the gross combination weight, the driver can always optimise driv-ability by selecting or deactivating the heavy duty mode, and activating the long haul mode. The functions in the software package also offer benefits for trucks hauling multiple trailers.TP-DIST is tailored for conditions in the distribution transport segment.TP-CON is adapted for construction operations.TP-LONG is designed to rationalise long-haul transport.TP-HD is specially tailored for heavier transport operations.• Standard (•) The function can be used when TP-LONG is activated. o Option – Not available *Only AT2612D, AT2612F, ATO2612F, ATO3112F and ATO3512F.†TP-BAS and TP-LONG are the only options available for SPO2812.Sales codes for I-Shift software packagesTP-BAS I-Shift basic software packageTP-DIST I-Shift distribution gear changing software TP-CON I-Shift construction gear changing software TP-LONG I-Shift long haul gear changing softwareTP-HD I-Shift heavy duty gear changing software Sales codes for standard equipmentAPF-BASS tandard version of I-Shift, if APF-ENH isnot chosenAMSO-BAS S tandard version of I-Shift, if AMSO-AUT isnot chosenAVO-BAS S tandard version of I-Shift, if AVO-ENH isnot chosen Sales codes for available optionsAPF-ENH Enhanced I-shift PTO functions (Auto N eutral /Reverse Inhibit / Split Box Connection)AMSO-AUT I-Shift manual gear shift available inautomatic mode incl kickdown functionAVO-ENH Enhanced I-Shift software for constructionand off road applicationsI-Shift’s software packages can easily be installed and changed with the help of Volvo’s analysis and programming tool, Volvo Tech Tool. This is done by authorised dealers and workshops, where the software packages can be further customised with optional functions and customer parameters.Basic PTO Functions (APF-BAS)Facilitates power take-off operation. Pre-defined splitter gear positions determine which splitter gear is used when one or two gearbox power take-offs are engaged.Because gear selection is matched to the engine speed limit, it is possible to set parameters for the software. The gear selection is then adapted to any engine speed limits imposed by body-builder functions.Enhanced PTO Functions (APF–ENH)Several functions that aid power take-off operation. I-Shift’s power take-off functions make it possible to activate the proper-ties listed below by having the software parameters adjusted at an authorised workshop.Auto NeutralOn command, the driveline is disconnected from the bodybuilder control unit, regardless of the gear lever’s position, when Auto Neutral is activated.Reverse InhibitWhen the bodybuilder control unit issues the Reverse Inhibit command, the reverse gears are blocked by the transmission system.Connection of splitter boxAllows connection of a splitter box for operation of high-capacity power take-offs. Direct gear is activated when the bodybuilder module is put in splitter box mode. It is also possible to use all high range gears. Please look into the body builder instructions.Basic Gear Selection Adjustment (AMSO-BAS) Allows the driver to adjust gear selection with the gear lever buttons during engine braking in Automatic mode (gear selec-tor position A).Enhanced Gear Selection Adjustment, incl. Kickdown (AMSO–AUT)This function allows both the automatically selected starting gear and the driving gear in Automatic mode to be adjusted by activating the plus/minus button on the gear lever. Arrow sym-bols in the driver information display show the available gears. There is also a function that facilitates speed adjustment when the vehicle is idling or driving very slowly, for instance in traffic queues. The gears can also be shifted upwards since engine speed is automatically increased before upshifts. The kickdown function selects a gear for maximum accelera-tion. When the kickdown switch on the accelerator pedal is engaged, the system changes the gearshift strategy to maximise vehicle acceleration. When suitable (e.g. depending on engine speed), this leads to a downshift.K ickdown only works in Economy mode to prevent accidental activation during off-road driving.Basic Vocational Functions (AVO-BAS)Allows the driver to choose between the Economy and Perfor-mance driving modes.Enhanced Performance – Bad Roads (AVO–ENH) This optional package is specially adapted to the specific conditions of the construction and timber transport segments. The P+ Performance mode includes various functions that adapt gearshifts and gear selection to poor driving surfaces and hilly gradients. It also includes functions that facilitate starting from standstill in poor driving conditions.P + is designed to minimise the number of gearshifts required. This is useful during off-road driving. It prevents wheels from spinning out when torque is increased after a gearshift, and prevents missed gearshifts, for example if the road gradient changes sharply. High engine power (high revs) is often required when driving uphill.If the driver speeds up before a hill and then changes gears, the truck may not gain enough speed.The driver can also influence the maximum number of downshifts.This is very useful when you shift to a lower gear on a very steep uphill gradient and only want to shift once to a gear strong enough to take you all the way up. Both Economy, Performance an P+ are now available.Summary of the functions in the package:• Engine revs are increased as necessary to provide extra torque when starting off from standstill.• Larger margins before upshifts ensure safer driving if the gradient changes.• Gear selection is adapted to minimise the number of gear-shifts and run at slightly higher revs (also available with Economy mode).• Functions that make it easier to keep the same gear when the• accelerator pedal position and road gradient change.• The package enables multiple downshifts. This facilitates gearshifts when driving up steep slopes.• Includes a function that speeds up clutch release and makes it easier to rock the vehicle out of trouble if it gets stuck on a soft surface.• When moving the gear lever, the driver can choose the gear that provides the highest possible engine speed.AVO-ENH can be combined with the Heavy Duty Transport program (TP-HD) without any problem. The AVO-ENH func-tions will only be active when the HD-mode is not active and when the Power mode is selected.Basic Shift StrategyAutomatic selection of correct starting gear (1st – 6th gear). The choice of starting gear is determined by gross vehicle weight and road gradient.Performance ShiftGives faster, gentler shifts through intelligent utilisation of the engine’s compression brake (VEB brakes), the vehicle’s clutch and a special gearbox brake.Gearbox Oil Temperature MonitorContinuously shows the gearbox oil temperature in the infor-mation display.Heavy Start EngagementFor start-up with high revs in Performance mode in 1st gear, resulting in higher starting torque. This function raises the revs to facilitate heavy starts. This is useful, for instance, if the truck is stuck in soft ground.I-RollAutomatic activation and deactivation of a freewheel function in order to cut fuel consumption, which can be reduced by up to several percent. I-Roll is used when neither engine power nor engine braking is needed, for instance on flat roads. When driving with cruise control, I-Roll runs at roughly 1–3 km/h be-low the pre-set speed, which saves fuel. The longer the vehicle drives using I-Roll, the more fuel is saved.Smart Cruise ControlInteracts with the vehicle’s Brake Cruise and ensures that the auxiliary brakes are not activated unnecessarily. The auxiliary brakes are deactivated on downhill stretches to save fuel. This allows increased use of the freewheel function, resulting in improved fuel efficiency.Launch Control4Optimises gear selection and EBS functions when manoeuvring at low speeds. Manoeuvring is facilitated because the EBS brakes are automatically engaged when the truck changes direction. This also ensures that the Hill Start Aid function is only activated on uphill gradients.It is possible to drive the vehicle forward with the idle regulator. This saves unnecessary downshifts and makes it easier to adjust the vehicle’s speed, for instance when driving in traffic queues. Enhanced Shift Strategy1By interacting with EBS2 and ECS3, both starting and ma-noeuvring are made easier.This brake mode maximises VEB/VEB+/retarder braking effects by automatically selecting the appropriate gear so the engine runs at high revs. This function compensates for the engine brake when changing gears in brake mode.When changing gears during engine braking, the wheel brakes are activated to compensate for braking moment. This raises braking power and provides smoother gearshifts.Interaction with the braking systems increases safety by preventing the truck from accelerating during gearshifts on steep slopes when braking mode is activated.Heavy Duty GCW Control5Optimises gear selection for high gross combination weights (85 t < GCW ≤ 180 t). This function improves driveability and fuel economy in the heavy duty transport segment. Heavy Duty GCW Control gives the driver access to the HD (Heavy Duty) driving mode.In HD mode, 1st gear is used as the starting gear and gear selection is adapted to heavier gross combination weights. The gearshifts generally occur at higher revs. HD is activated and deactivated by pressing and holding the E/P button on the gear selector for about 3 seconds. The chosen driving mode remains selected when the engine is turned off.Among other things, the TP-HD function selects the start-ing gear to suit the gross combination weight, thereby saving the clutch. The entire gear range is utilised, and the gears are changed consistently at high revs to maintain torque and driving comfort.When driving with low gross combination weights or without a load, it is easy to deactivate the HD driving mode and return to Economy mode. After this, the driver can switch between Economy and Performance modes. This ensures comfortable and fuel-efficient driving.1 Full functionality requires EBS-MED.2 EBS = Disc Brakes with Electronically controlled Brake System (EBS-STD / EBS-MED)3 ECS = Electronically Controlled Suspension (SUSPL-EC).4 Full functionality requires EBS and ECS.5 Available only with certain engine/gearbox combinations. Customer parametersI-Shift also has many options for setting customer parameters that optimise the vehicle’s driving properties in special applica-tions and special transport segments. For instance, the starting gear can be optimised according to the transport conditions. Power take-off operation can also be customised. Customised settings and reprogramming of I-Shift are car-ried out at authorised workshops using the Volvo Tech Tool.Volvo retains the right to modify design and specifications without prior notification.。

361 Chapter 14Techniques for dye injection and cell labelling PETER MOBBS, DA VID BECKER, RODDY WILLIAMSON, MICHAEL BATE and ANNE WARNER1. IntroductionThe introduction of compounds into cells via iontophoresis or pressure injection from micropipettes is a powerful technique of wide application in modern biology. The many uses to which this technique can be put include:(i) Cell identification following electrophysiological recording.(ii) Delineation of cellular architecture in anatomical studies.(iii) Tracing neuronal pathways.(iv) Identification of cell progeny in lineage studies.(v) Investigations of the transfer of molecules from one cell to another via gap junctions or other routes.(vi) The introduction of genetic material that affect protein synthesis or gene expression.(vii) The measurement of intracellular ion concentrations, for example pH or calcium ion.This chapter describes the techniques used to inject cells and focuses upon the design of experiments for some common applications of these methods. In the final sections, we offer sample protocols and advice on the necessary equipment.The basic methods for cell injection are similar whatever the compound to be used. This chapter concentrates on techniques that involve iontophoresis or pressure injection using intracellular micropipettes while section 9 describes some other routes by which compounds can introduced into cells. For each application described below, we concentrate upon the factors that influence thePETER MOBBS, Department of Physiology, University College London, Gower St., London WC1E 6BT, UKDAVID BECKER AND ANNE WARNER, Department of Anatomy and Developmental Biology, University College London, Gower St., London WC1E 6BT, UKRODDY WILLIAMSON, The Laboratory, Citadel Hill, Plymouth, PL1 2PB, UK MICHAEL BATE, Department of Zoology, University of Cambridge, Downing St., Cambridge CB2 3EJ, UK362P. MOBBS AND OTHERSchoice of the compound to inject, since this is usually the factor most crucial to success.2. Microinjection methodsManufacturing micropipettesPipettes for intracellular microinjection can be produced on any standard microelectrode puller. The best pipettes generally have the following characteristics: (a) a relatively short shank (b) a relatively large tip diameter. The latter is frequently a limitation because, for successful penetration of small cells without damage, the tip diameter also must be small. When the diameter of the tip is small then both the iontophoresis and pressure injection of compounds is impeded, the former by the charge on the glass and the electrical resistance of the tip and the latter by the tip’s resistance to bulk flow of solution. Several different types of glass are available for the production of micropipettes. A number of manufacturers (see appendix B) provide suitable capillaries with a variety of outside diameters, with thick or thin walls, with and without internal filaments, made from soda or borosilicate glass. Pipettes made from thick-wall borosilicate glass are usually the most robust and useful for penetrating tough tissue. However, thin-wall glass has the advantage that the channel through the tip is usually larger, and thus the resistance is lower, for any given tip size. The characteristics of micropipettes for use in microinjection experiments can sometimes be improved by bevelling (see Chapter 11). Soda-glass is somewhat less fragile than borosilicate glass but is difficult to pull to fine tips, it has been dropped from some supplier’s lists. No matter what the theoretical expectations, the best electrodes to use are those that work!Pipette fillingModern micropipette glass incorporates an internal ‘filament’ (actually a second narrow capillary). The filament increases the capillarity of pipettes so that fluid is drawn into the tip. This characteristic can be exploited to enable very small volumes of fluid to be loaded into the pipette tip, which is useful where the compound to be injected is expensive. Solutions can be introduced into the back of the pipette either by immersion or by bringing into contact with a drop of fluid. The volume drawn into the tip depends upon its diameter. Pipettes with tips of 1 µm will draw up about 100 nl and those of 5 µm will fill with about 1 µl of fluid. Coarse pipettes can be filled by sucking fluid directly through the tip. Electrical connections to pipettes in which only the tip is filled can usually be effected simply by sticking a wire into the pipette lumen. The presence of a thin trail of electrolyte along the outside of the internal filament provides the necessary path for current flow. It is advisable to centrifuge all solutions before use to remove material that may block the tip.IontophoresisIontophoresis involves the ejection of a substance from a pipette by the application ofcurrent. The polarity of the ejection current employed depends on the net charge on the substance to be injected (negative pulses are used to eject negatively charged molecules). Most modern microelectrode amplifiers are equipped with a current pump that can be used to provide an iontophoretic current that is, within limits,independent of the electrode resistance (see Chapters 1 and 16). If only a simple amplifier is available, or the current pump is unable to provide sufficient voltage to drive the required current through the electrode tip, then it is possible to use a battery and a current limiting resistor as a current source. If a battery is employed then the headstage of the amplifier should be switched out of the circuit when the battery is connected. Obviously the current provided by this crude arrangement will be governed by Ohm’s Law. The current applied to a cell should be as small as is consistent with the introduction of sufficient of the compound into the cell. In all events the voltage produced by the passage of the iontophoretic current must be limited (to say +100 to −100 mV) to avoid damage to the cell membrane.Continuous application of current should be avoided since it often causes the electrode tip to block. This block can sometimes be relieved by reversing the polarity of the current for a short time. However, once an electrode shows signs of block the trend is usually irreversible and the pipette should be discarded. Often the best strategy is to employ short duration current pulses of alternating polarity. Whatever the form of the pulse, small currents for long periods are usually more successful than high currents for shorter times. To recognise electrode block and standardise procedures, it is essential to monitor the current flow through the electrode. It is not sufficient simply to monitor the voltage applied to the electrode! If the amplifier employed does not have a current monitor then a simple one can be improvised by measuring the voltage drop across a resistor in the earth return circuit. The membrane potential of the cell should be measured during electrode insertion, before switching to current injection. It is sensible to check the condition of the cell by measuring its resting potential at intervals during iontophoresis. Such measurements are simplified by using a bridge amplifier (see Chapters 1 and 16) that enables the membrane voltage to be monitored continuously during current passing experiments. For a detailed discussion of the circuits for current injection and current monitoring see Purves (1981).A useful technique for achieving bulk flow from the electrode tip is to cause high frequency oscillations of the voltage across the electrode resistance. This is achieved by pressing the ‘buzz’ or ‘zap’ buttons present on some amplifiers. The effect of these can be imitated by turning up the capacity compensation control, found on nearly all microelectrode amplifiers, to the point at which the electrode voltage oscillates (termed ‘ringing’).In theory the amount of a substance ejected from the pipette during an iontophoretic pulse can be estimated from a consideration of its transport number (Purves, 1981). In practice, these estimates are highly unreliable and the transport number is often unknown for the compound employed.Pressure injectionPressure ejection is the method of choice for the injection of neutral molecules and 363Techniques for dye injection and cell labelling364P. MOBBS AND OTHERSthose of low iontophoretic mobility. Commercial pressure injection devices are available (see list of suppliers) that enable the application of calibrated pressure pulses to the back end of the injection pipette. Essentially a pressure injection system consists of a gas cylinder connected, via a timing circuit, a solenoid-operated valve and a pressure regulator, to a side-arm pipette holder. Commercial equipment is expensive, but a home-made rig can be simply made from the components listed above. The timing circuit can be replaced by a manually operated switch. Take care to ensure that the connections and tubing are safe at the pressures employed and that the pipette is firmly held within the holder. The pressure and timing of the pulse can be roughly established by measuring the diameter of a drop expelled from the pipette tip into a bath of liquid paraffin. However, this method frequently over-estimates the back-pressure from the cytoplasm and quantification of pressure injection is often as uncertain as in iontophoresis.Patch-pipettesMany substances can be introduced into cells from patch-pipettes while recording in the whole-cell mode. The concentration that a compound reaches within the cell during whole-cell recording is equal to that within the patch-pipette solution. Thus for most dyes and labels the concentrations to employ are a fraction of those used in iontophoresis or pressure injection experiments. For example, Lucifer Yellow CH incorporated into the patch-pipette solution at 1 mg ml−1will produce intense fluorescence of the cell (40 mg ml−1is used in sharp electrodes for iontophoresis; Fig. 1B).3. Techniques for visualizing cellsVisualizing cells prior to injectionIn order to inject a cell you must be able to guide your micropipette toward it. There are three techniques available to aid in the steering of electrodes:(a) Stereotaxic movements combined with continuous electrical recording (mainly used for penetration of cells in brain nuclei).(b) Visual guidance using white light and interference contrast optics to visualize the cell and identify targets.(c) Visual guidance using cells prelabelled with fluorescent dyes as the target.In solid tissue, whatever technique is chosen to guide the electrode, the target must lie along initial trajectory of the electrode. Manipulation out of this axis will break the electrode.1. Stereotaxis. This method requires that you know precisely where your target cells lie even though you can not see them. Such information is sometimes available from stereotactic atlases. Micromanipulators can be roughly calibrated to give depth measurements but errors always arise as a result of tissue distortion during electrode penetration. The identification of the target cells can sometimes be achieved through knowledge of their electrical properties or synaptic connections,for example by the response to current injection or stimulation of a peripheral nerve.2. Interference contrast optics.Phase contrast and differential interference contrast techniques (Nomarski) are good for visualising living cells. Phase contrast is useful for cells in tissue culture but does not work well for tissue slices. Nomarski optics provide high resolution and can be used to provide effective optical sections of transparent tissue. The more recently introduced Hoffman optics are cheaper than Nomarski optics and are useful for viewing tissue slices because they provide a greater depth of field.3. Prelabelling with a fluorophore.There are two approaches to the prelabelling of cells to identify them as targets for subsequent microinjection experiments. Cells can either be bathed in a dye that becomes internalized (Fig. 1E), or labelled by retrograde transport of a marker from their axons (Fig. 1A). Whilst some dyes are either actively taken into cells or simply diffuse across the membrane others only enter if the membrane is disrupted by osmotic shock or through exposure to dimethyl sulphoxide. Whatever the method of prelabelling, the choice of the label is crucial to success. Ideally the label should be visible under the same filter set as the dye used in subsequent injection experiments and the intensity of the prelabel’s fluorescence should not mask that of the injected fluorophore. Since the prelabel may remain inside the cell for an extended period, it is important that it is non-toxic.Retrograde labelling of neurons via their axonal projections is an extremely useful means of identifying populations of cells that project to particular targets. Fast blue and diamidino yellow are amongst the most popular of the labels available for this purpose. Fast blue labels the cell cytoplasm and diamidino yellow stains the nucleus (Fig. 1E). Both pass rapidly across the cell membrane and can be used to label cells from their axon terminals or from cut axons. General labelling of all the cells in a tissue can be achieved by bathing in a dilute solution of the dyes. Both of these dyes work well on formaldehyde-fixed tissue. Target cells identified with these prelabel dyes can subsequently be injected with Lucifer Yellow, carboxyfluorescein or Cascade Blue which are visible with the same filter set (Fig. 1E).Some fluorophores with useful properties are neither taken up nor transported by cells. However, they can be made into useful labels through conjugation to lectins,dextrans or plastic microspheres. Lectins bind to sugar moieties on the cell membrane, are brought into the cell through endocytosis and transported. Dextrans can also be conjugated to most fluorophores. Plastic microspheres can be coupled to fluorescent molecules. They are available in a variety of materials and sizes. Applied to damaged axons they are taken up by and retrogradely transported. Microspheres are visible in the electron microscope.Visualizing labelled cellsThe object of many microinjection experiments is to render the cell under study visible by introduction of a label. The majority of such labels are either fluorescent or can be processed to produce a coloured reaction product. Below we describe the techniques for visualizing and recording the results of cell labelling experiments.365Techniques for dye injection and cell labelling366P. MOBBS AND OTHERSOften labelled cells can be visualised without any histological processing and some labels can be used to follow changes in cell morphology that occur over extended periods of time (Purves et al.1986). Methods for the fixation of tissue and the histological processing of tissue containing labels are given later.Fluorescent labels are excited by light at one wavelength and emit light at another longer wavelength. The user must choose the excitation and emission filters most suitable to their application (see appendix A). It is convenient to have the microscope used for positioning the electrode equipped with a light source and filters capable of exciting the label. This allows the user to determine the endpoint of the injection experiment by observation. Many of the labels in common use are excited by far blue or UV light. The tungsten or quartz halogen bulbs found in most microscope illuminators do not provide much light at these wavelengths and an additional mercury or xenon light source is required. Most manufacturers provide some convenient means for switching between the white and UV light sources. If this switching mechanism is to be used during the course of a labelling experiment, it is imperative that it operates without vibration if the microelectrode is to remain in the cell under study.The factors involved in the choice of the optics and light sources for fluorescence microscopy are complex. The short discussion below is offered as an introduction that may be supplemented by consulting some of excellent free literature provided by major manufacturers (see for example the booklets offered by Zeiss, Lieca and Nikon). Mercury lamps are cheaper than xenon lamps. However, the emission spectrum of a xenon lamp is relatively continuous throughout the UV and visible spectrum while that of mercury lamps consists of a series of sharp peaks (emission lines). With mercury lamps, it is important to ensure that a line exists at a wavelength appropriate to the dye in use. Most modern fluorescence microscopes employ epi-illumination, a system in which the light used to excite the dye is focused on the specimen through the same objective used to view the light emitted by the dye.The choice of objective is critical in fluorescence microscopy. Quartz objectives pass much more short wavelength light than those made from glass. However, quartz objectives are expensive and unnecessary for use with dyes excited by light in the visible and near UV regions of the spectrum. It is crucial that the objective has a high numerical aperture (NA) since both the intensity of the light focused on the specimen and the light gathering power of the lens increase with the square of the aperture. An objective with an NA of 1.0 will yield 16 times as much light as a 0.5 NA lens. High NA objectives have shorter working distance and need an immersion medium - water, oil or glycerol (for UV). For injection of cells in thick preparations on an upright microscope water immersion lenses are preferable to those that work in air because they have a greater NA and there is no optical distortion due to meniscus effects of the micropipette on the bath surface. On the other hand, very long working distance air electrodes can be covenient, if optically inferior. Two particularly useful lenses are Zeiss ×40 0.75 NA W water immersion and the Nikon ×40 ELWD air (NA 0.5) with correction collar. Intensity of fluorescent light also depends upon the magnification. Itdecreases as the square of the magnification: a ×10 eyepiece produce an image of 25% the intensity of an image formed by a ×5 eyepiece. Low magnification eyepieces are therefore preferable for visual observation.Fluorescent images can be recorded on film or by analog or digital video techniques. There are many black and white, colour print and transparency films suitable for recording fluorescence images. Generally a film of high speed and acceptable grain should be chosen. Colour films of speed greater than 400 ASA tend to be too grainy, however, black and white films such as Kodak’s TMAX give excellent results even at 2400 ASA (must be developed in TMAX developer). In normal photography, the reciprocity law applies and the total amount of exposure is given by the product of the luminance and the exposure time. Thus an exposure of 1/60th of a second at f8 is the same as for 1/30th at f11. With dim objects the reciprocity law fails to predict the exposure and the exposure time has to be increased.Most film manufacturers provide a guide to the performance of their films at low light intensities. In practice it is often better simply to take several exposures of increasing duration starting with the exposure time indicated by the meter on the camera.The advent of cheaper video cameras that operate at low light intensities has opened up the possibility of recording fluorescent images either on video-tape or in digital form on a computer. Digital image recording has the advantage of allowing complex analysis of an image.Labels that result in a coloured or opaque reaction product are much simpler to photograph than those labelled with fluorescent compounds. No special equipment is required.4. Labelling cells for subsequent identification and fordetermination of overall cell architectureDyes injected for these purposes should have the following properties: (a) they should be visible, either immediately or after chemical reaction; (b) they should remain in the injected cell, either because they are too large to move across the cell membrane and through gap junctions or because they are strongly bound by the cytoplasm; (c) they should not be toxic, although this requirement can be relaxed if the tissue is to be processed immediately after the cell has been injected; (d) they should be stable and not break down to give products with different properties; (e)they should withstand histological processing. In practice, property (e) is the most difficult to achieve.Six classes of compound are used for this purpose:1. Inherently fluorescent molecules and those tagged with a fluorescent probe.Lucifer Yellow (MW 457) and carboxyfluorescein (MW 376) are the most popular fluorescent compounds for determining overall cellular architecture. However, they are far from ideal for this purpose. Both pass through gap junctions (see below) and carboxyfluorescein cannot be fixed. Lucifer Yellow withstands fixation well but as 367Techniques for dye injection and cell labelling368P. MOBBS AND OTHERSwith all other dyes some fluorescence intensity is lost. Passage through gap junctions can be prevented by conjugation of the fluorophore to dextrans. Dextrans (MWs 3000-70000) can be coupled to fluorescein, rhodamine isothiocyanate or Texas Red. They can be prepared in the laboratory (see Gimlich & Braun, 1985) or purchased commercially (Molecular Probes, 48-49 Pitchford Avenue, Eugene, Oregon, OR97402-9144 USA). Cascade Blue and sulphrhodamine 101 are also useful for determining cellular architecture and extend the range of colours available for double marking experiments. For examples of multiple labelling see Fig. 1D,F. Advantages:Can be pressure injected or iontophoresed.Can be seen in living cells with appropriate fluorescent illumination.Are not toxic provided the amount injected is kept fairly low.Do not break down.Will withstand routine fixation and embedding techniques, provided the fixative or mountant does not generate auto-fluorescence. Glutaraldehyde fixation, for example, must be avoided. Many commercial mountants, such as DPX, are unsuitable for this reason. Mountants that are designed to reduce fading can now be obtained (e.g. Citifluor, City University, London). Disadvantages:Limit of detection determined by threshold of fluorescence. Detection levels can be improved by electronic image intensification.Fluorescence fades under continuous illumination. This can be reduced by using anti-fade mountants.Fluorescein fades particularly fast, but is more fluorescent than rhodamine or Texas Red.Sometimes become incorporated into cellular organelles with time, making fluorescence particulate.Margin between visible not toxic, and visible but toxic is narrow.2. The carbocyanine dyes.Octadecyl(C18)-indocarbocyanine (DiI) and oxycarbocyanine (DiO) (MWs 934 and 882) are highly fluorescent lipophilic compounds. They dissolve in, and diffuse throughout, the lipids of the plasma membrane. They are not toxic and they have been reported to remain in the cell membrane for up to one year (Kuffler, 1990). They will also diffuse along membranes in lightly fixed tissue. In the absence of any sites of membrane fusion the carbocyanines label single cells. The diffusion rate for these compounds is slow (about 6 mm/day, slower in fixed tissue), however, carbocyanines with unsaturated alkyl chain segments (FAST-DiI and FAST-DiO) exhibit accelerated diffusion rates. The polyunsaturated “DiASP” compounds (N-4(4-dilinoleylaminostyryl)-N-methylpyridinium iodide and related molecules) (MW~800) are also reported to diffuse more rapidly. Because the carbocyanines are insoluble in water they must either be pressure injected into cells in solution inDMSO or alcohol or applied to the cell membrane in which they rapidly dissolve.DiI and DiO can be visualized by fluorescence microscopy. DiI has similar excitation properties to rhodamine, excited by green it fluoresces red. DiO is similar to fluorescein in that it is excited by blue light and produces green fluorescence. DiAsp has a broad excitation spectrum and fluoresces orange. These dyes can be converted into a permanent reaction product via the Maranto reaction (Maranto, 1982) in which the singlet oxygen released by illumination is used to oxidise diamino-benzidine (DAB).Advantages:They are not toxic and can remain in the cell membrane without harm over several years.Disadvantages:Not water soluble.They tend to fade quickly particularly in laser scanning confocal microscopy.Long diffusion times.Can only be pressure injected.3. Enzymes such as horse radish peroxidase. Horse radish peroxidase (HRP) is reacted with diamino-benzidine or other chromogens to generate a product visible in the light or electron microscope. There are many protocols for developing HRP (see Mesulam, 1982 and Heimer & Robards, 1981 for a selection). Widely used in studies in the central nervous system. The injection of enzymes can also be used to kill individual cells (e.g. pronase). This is potentially useful in lineage and regeneration studies.Advantages:Can be pressure injected or iontophoresed.Not toxic.Remains within the injected cell, provided the preparation is free from micro-peroxidases. Will cross synapses, which can be useful when tracing pathways.Does not break down.Good visibility.Reaction product visible in the electron microscope.Disadvantages:Can only be seen after reaction product produced. However, by using a fluorescent peroxidase conjugate, such as RITC-peroxidase (Sigma P5031),an indication of the staining can be obtained during the fill period (see Fig.1A-C).Can get reaction product from endogenous peroxidases, so method has to be modified if this is likely to be a problem.The penetration of chromogen into tissue is rather poor (about 100 µm), so that whole mounts or slices have to be below this thickness.369Techniques for dye injection and cell labelling370P. MOBBS AND OTHERSMuch of the enzyme activity is lost on fixation. If possible the material is best fixed after reaction.4.Biocytin. A recently introduced intracellular marker (Horikawa & Armstrong, 1988) comprising a highly soluble conjugate of biotin and lysine (MW 372.48) that has a high binding affinity for avidin. The injected biocytin is visualised by attaching a label to avidin, e.g. a fluorescent label such as FITC or rhodamine, or a chromogenic enzyme such as HRP. Suitable avidin conjugates are widely available (e.g. Sigma, Vector Labs.). A small molecular weight biotin compound, biotinamide (MW 286), is also available (Neurobiotin, Vector Labs, 16 Wulfric Square, Bretton, Peterborough PE3 8RF, UK) and may be easier to inject (Kita & Armstrong, 1991). Advantages:Highly soluble in aqueous solutions.Can be pressure injected or iontophoresed.Low toxicity.Does not break down.Good fluorescent, visible light, or electron microscopic visibility after avidin reaction.Disadvantages:Can only be seen after avidin reaction.Reaction penetration limited to about 100 µm even with detergents or surfactants so tissue may have to be sectioned.Some ultrastructural degradation from penetration agents.Can pass between coupled cells.Occurs naturally in trace amounts.5. Heavy metals such as cobalt and nickel.The metal is precipitated with ammonium sulphide or hydrogen sulphide. The sensitivity can be improved by intensification with silver (Pearse, 1968; Bacon & Altman, 1977). Double labelling can be achieved by using different metals in the same preparation followed by precipitation with rubeanic acid (Quicke & Brace, 1979); this results in precipitates of different colours depending on the metal, e.g. cobalt = yellow, nickel = blue, copper = olive.Heavy metal complexes, such as lead EDTA (Turin, 1977) can be suitable in cells that are not linked to their neighbours by gap junctions (see later section). In principle, it is possible to prepare a range of heavy metal complexes of different sizes so long as the complex is firmly held, so that there is no free metal or anion which might be toxic, and the metal has a much higher affinity for sulphide than for the anion used to make the complex. This is essential to ensure precipitation of the metal out of the complex. The advantage of a heavy metal complex is that the complex can be much less toxic than the heavy metal itself and may be much easier to eject from the pipette. However, some metal sulphides will re-dissolve if the precipitant (usually ammonium sulphide) contains polysulphides. Freshly prepared solutions saturated with H2S do not suffer from polysulphide formation.。

Mini Trans-Blot®Electrophoretic Transfer Cell Instruction ManualCatalog numbers170-3930170-3935170-3989170-3836Assembly and DisassemblyTo insure best performance from the Mini Trans-Blot®electrophoretic transfer cell, become fully acquaintedwith these operating instructions before using the cellto transfer samples. Bio-Rad recommends that you first read these instructions carefully. Then assemble and disassemble the cell completely. After these preliminary steps, you should be ready to transfer a sample.Wash Cell Before UseBio-Rad also recommends that all Mini Trans-Blot electrophoretic transfer cell components and accessories be cleaned with a suitable laboratory cleaner (such asBio-Rad Cleaning Concentrate, catalog #161-0722) and rinsed thoroughly with distilled water before use. WarrantyBio-Rad Laboratories warrants the Mini Trans-Blot electrophoretic transfer cell against defects in materials and workmanship for 1 year. If any defects occur inthe instrument during this warranty period, Bio-Rad Laboratories will repair or replace the defective parts free. The following defects, however, are specifically excluded:1. Defects caused by improper operation.2. Repair or modification done by anyone other thanBio-Rad Laboratories or an authorized agent.3. Use of fittings or other spare parts supplied by anyoneother than Bio-Rad Laboratories.4. Damage caused by accident or misuse.5. Damage caused by disaster.6. Corrosion due to use of improper solvent or sample. For any inquiry or request for repair service, contactBio-Rad Laboratories after confirming the model and serial number of your instrument.Mini-Trans-Blot Electrophoretic Transfer Cell iTable of ContentsAssembly and Disassembly (i)Wash Cell Before Use (i)Warranty (i)Section 1 Introduction (1)1.1 Specifications (3)1.2 Safety Instructions (4)Section 2 Mini Trans-Blot Cell Assembly and Preparation for Transfer (5)2.1 Mini Trans-Blot Cell Description andAssembly of Parts (5)2.2 Preparation for Blotting (6)2.3 Acidic Transfers (9)Section 3 Transfer Conditions (10)3.1 General Guide to Transfer Buffers andRunning Conditions (10)3.2 Notes on Electrophoretic TransferConditions (11)3.3 Buffer Formulation (13)Section 4 Strategies for OptimizingElectrophoretic Transfer (15)4.1 Optimizing Protein Transfer (15)4.2 Optimizing DNA and RNA Transfer (18)Section 5 Choice of Blotting Membranes (19)5.1 Protein Blotting (19)5.2 DNA and RNA Blotting Membranes (20)Section 6 Troubleshooting Guide (22)6.1 Electrophoretic Transfer (22)Section 7 References (27)Section 8 Product Information (29)Section 1IntroductionBlotting was first performed by Southern in 1975 withthe transfer of DNA from agarose gels to nitrocellulose membranes.1 Since that time, blotting has been applied to RNA2-4 and proteins5, 6 in both agarose and polyacrylamide gels. To circumvent the inefficiencies observed in various capillary transfers, electric current has been adopted for eluting proteins from polyacrylamide gels,as first described by Towbin et al. in 1979.7 The use of electrophoretic transfer has also been applied to DNA and RNA blotting.8–14 Numerous publications have dealt with the topic of protein electrophoretic transfer techniques.15–26 There have also been reviews summarizing the expanding literature being generated on electrophoretic blotting methodology.27–29The Mini Trans-Blot® tank is part of Bio-Rad’s modular Mini-PROTEAN® Tetra system. The unique feature of this electrophoresis system is that the electrode modulesare interchangeable. After finishing gel electrophoresis, remove the electrode module from the buffer tank, insert a new electrode module, add new buffer, and the next electrophoresis application can be performed.The Mini Trans-Blot module accommodates two cassettes for electrophoretic transfer. The Mini Trans-Blot module is useful for blotting either protein or nucleic acid from both agarose and acrylamide gels. It is also capable of blotting isoelectric focusing gels from horizontal electrophoresis cells, or DNA and RNA gels from the Mini-Sub® submarine electrophoresis cell. For applications where the gel is larger than 7.5 x 10 cm, or when there are more than two mini gels to be transferred, the larger standard Trans-Blot®cell (catalog #170-3910 or 170-3946), Criterion™ Blotter (catalog #170-4070, 170-4071) or the Trans-Blot® SD semi-dry cell (catalog #170-3940) should be used.The heart of the Mini Trans-Blot cell is its electrode module. This module has the capacity to hold two gel cassettes between parallel electrodes only 4 cm apart. The driving force for blotting applications is the voltage applied over the distance between the electrodes.Mini-Trans-Blot Electrophoretic Transfer Cell 1This short 4 cm electrode distance allows generation of higher driving forces to produce efficient protein transfers.A second feature of the electrode module is that it is offset to accommodate a blue cooling unit. The cooling unit, which is completely contained within the Mini Trans-Blot cell, absorbs the Joule heat generated during rapid electrophoretic transfers. The advantages of having an internal cooling unit include elimination of an expensive external cooling bath and avoidance of cumbersome cooling tubing. Other features of the Mini Trans-Blot cell include gel holder cassette latches for easy handling, color coordinated cassettes and electrodes to insure proper orientation of the gel during transfer, and an efficient design which simplifies insertion and removal of the cassettes from the electrode assembly. These features result in an electrophoretic transfer system which is easy to use and produces excellent blotting results.2 Mini-Trans-Blot Electrophoretic Transfer Cell1.1 SpecificationsConstructionElectrode module Molded polysulfoneGel holder cassettes Molded polycarbonateElectrodes Platinum wire 0.254 mmdiameterBuffer chamber and lid Molded polycarbonateCooling unit PolyethyleneOverall dimensionsMini Trans-Blot cell16 (L) x 12 (W) x 18 (H) cmGel holder dimensions10 x 11 cmMaximum gel size7.5 x 10 cmBuffer capacityWith cooling unit950 mlWithout cooling unit1,150 mlCleaning Use mild soap and warmwater to clean the electrodes,cassettes, and buffer tank.Use special care whencleaning the electrode cards.Avoid stretching or breakingthe platinum wires. Do notuse abrasives or strongdetergents. Rinse the fiberpads under hot water andthen in distilled, deionizedwater.Chemical compatibility The Mini Trans-Blot cellcomponents are notcompatible with chlorinatedhydrocarbons (e.g.,chloroform), aromatichydrocarbons (e.g., toluene,benzene), or acetone. Use oforganic solvents voids allwarranties.Mini-Trans-Blot Electrophoretic Transfer Cell 34 Mini-Trans-Blot Electrophoretic Transfer Cell 1.2 Safety InstructionsPower to the Mini Trans-Blot cell is supplied by an external DC voltage power supply. This power supply must be ground isolated in such a way that the DC voltage output floats with respect to ground. All of Bio-Rad’s power supplies meet this important safety requirement. Regardless of which power supply is used, the maximum specified operating parameters for the cell are:400 VDC Maximum voltage limit500 W Maximum power limit 40°CMaximum ambient temperature limit Current to the cell, provided from the external powersupply, enters the unit through the lid assembly,providing a safety interlock to the user. Current tothe cell is broken when the lid is removed. Do notattempt to circumvent this safety interlock, andalways turn the power supply off before removingthe lid, or when working with the cell in any way.Important : This Bio-Rad instrument is designed and certified to meet IEC61010-1 and EN61010-1* safety standards. Certified products are safe to use when operated in accordance with the instruction manual. This instrument should not be modified or altered in any way. Alteration of this instrument will:•Void the manufacturer’s warranty •Void the IEC61010-1 and EN61010-1 safety certification • Create a potential safety hazardBio-Rad is not responsible for any injury or damage caused by the use of this instrument for purposes other than for which it is intended or by modifications of the instrument not performed by Bio-Rad or an authorized agent.* IEC61010-1 and EN61010-1 are internationally accepted electrical safety standard for laboratory instruments.!!Section 2Mini Trans-Blot ® Cell Assembly and Preparation for Transfer2.1 Mini Trans-Blot Cell Description and Assembly ofPartsFilter paper LidFiber padMembraneGel Filter paper Fiber padElectrodemoduleBlue cooling(keep frozen at–20°C)Buffer tankGel holdercassette2.2 Preparation for BlottingStore the blue cooling unit in your laboratory freezer at–20°C until ready to use. After use, rinse the outside container with water and return the cooling unit to the freezer for storage.1. Prepare the transfer buffer. (See Section 3.3 for bufferformulation. Using buffer chilled to 4°C will improveheat dissipation.)2. Cut the membrane and the filter paper to thedimensions of the gel or use precut membranesand filter paper. Always wear gloves when handlingmembranes to prevent contamination. Equilibrate the gel and soak the membrane, filter paper, and fiberpads in transfer buffer (15–20 min depending on gel thickness).3. Prepare the gel sandwich.a. Place the cassette, with the gray side down,on a clean surface.b. Place one prewetted fiber pad on the grayside of the cassette.c. Place a sheet of filter paper on the fiber pad.d. Place the equilibrated gel on the filter paper.*e. Place the prewetted membrane on the gel.*f. Complete the sandwich by placing a piece offilter paper on the membrane.*g. Add the last fiber pad.* Removing any air bubbles which may have formed is very important for good results. Use a glass tube or roller to gently roll out air bubbles.4. Close the cassette firmly, being careful not to movethe gel and filter paper sandwich. Lock the cassette closed with the white latch.5. Place the cassette in module. Repeat for the othercassette.Fiber padFilter paperMembraneGel Filter paper Fiber pad6. Add the frozen blue cooling unit. Place in tank and fillto the “blotting” mark on the tank.7. Add a standard stir bar to help maintain even buffertemperature and ion distribution in the tank. Set the speed as fast as possible to keep ion distributioneven.8. Put on the lid, plug the cables into the power supply,and run the blot. Refer to Section 3 for run times and voltage settings with various buffers.9. Upon completion of the run, disassemble theblotting sandwich and remove the membrane fordevelopment. Clean the cell, fiber pads, and cassettes with laboratory detergent and rinse well with deionized water.2.3 Acidic TransfersIf transferring under acidic conditions, switch the gel and membrane in the set up instructions. This will place the membrane on the cathode side of the gel. Under acidic conditions, proteins will transfer in the opposite direction going toward the negative cathode.Section 3Transfer Conditions3.1 General Guide to Transfer Buffers and Running ConditionsTable 3.1 provides guidelines for power conditions using different buffers. Power conditions are provided for various run times. Where multiple conditions are displayed, the higher the voltage, the less time required for the run. Always use the blue cooling unit.Table 3.1. Guide to Buffers and Running Conditions.Buffer Standard Field HighIntensity FieldBuffer OvernightTransfer High Intensity Field 1 Hour TransferSDS-PAGE Gels Buffer A or B or C Buffer A or B or CA: 25 mM Tris, pH 8.3, 192 mM glycine, with or without 20% MeOH and .025%–0.1% SDS 30 V, constant90 mA100 V, constant350 mAB: 48 mM Tris, pH 9.2, 39 mM glycine, with or without 20% MeOH and .025%–0.1% SDS C: 10 mM NaHCO3, 3 mM NaCO3, pH 9.9, with or without 20% MeOH and.025%–0.1% SDSDNA and RNATAE: 20 mM Tris, pH 7.8, 10 mM 30 V, constant100 mA80 V, constant500 mATBE: 50 mM Tris, pH 8.3, 50 mM sodium borate, 1.0 mM EDTANative Gels25 mM Tris, pH 8.3,92 mM glycine. No methanol.30 V, constant90 mA100 V, constant350 mAIsoelectric Focusing, Native Gels, Basic Proteins, Acid Urea Gels*0.7% acetic acid30 V, constant100 mA 100 V, constant 350 mA*Please refer to Section 2.3 before transferring.3.2 Notes on Electrophoretic Transfer Conditions These variables will change total resistance and thus the current readings:• Alterations in buffer make-up, i.e., addition of SDS, or changes in ion concentration due to addition of acidor base to adjust the pH of the buffers• Gel pH, ionic strength, and percentage of acrylamide, especially if the gel has not been properly equilibrated • Number of gels; current increases slightly as the number of gels increases• Volume of buffer; current increases when volume increases• Platinum mass; current increases when mass increases• Transfer temperature; current increases when temperature increases• Time in transfer at which reading was taken;current normally increases as the buffering capacitydiminishes with progress of the runPre-equilibration of gels (15–20 min)All electrophoresis gels should be pre-equilibrated in transfer buffer prior to electrophoretic transfer.Pre-equilibration will facilitate the removal of contaminating electrophoresis buffer salts and neutralization salts (salts resulting from the denaturation of nucleic acids prior to transfer). If the salts are not removed, they will increase the conductivity of the transfer buffer and the amount of heat generated during the transfer. Also, low percentage gels will shrink in methanol buffers. Equilibration allows the gel to adjust to its final size prior to electrophoretic transfer. Current limitsThe PowerPac™ Basic power supply is capable of a75 W output. Unless a current limit is set, uncontrolled conductivity changes may result in full power being delivered to the Mini Trans-Blot® cell.The gel holders may warp, and the transfer buffer may boil and evaporate (further increasing conductivity). This would result in a potential safety hazard. Refer to the PowerPac Basic power supply instruction manual for setting current limits and run times. The Mini Trans-Blot cell is also compatible with the PowerPac HC power supply.Use of a stir bar during transferFor all blotting applications a stir bar must be placed inside the Mini Trans-Blot cell and the entire unit be placed on a stir bar mixer, so that the transfer bufferis stirred during the course of the experiment. This will help to maintain uniform conductivity and temperature during electrophoretic transfer. Failure to properly control transfer buffer temperature results in poor transfer of macromolecules and poses a potential safety hazard. Transfer buffer pHDo not adjust the pH of transfer buffers unless specifically indicated. Adjustments of the transfer buffers pH, when not indicated, will result in increased buffer conductivity. This is manifested by a higher than expected initial current output and a decreased resistance. It is recommended that the buffer conductivity and resistance be checked with the PowerPac Basic power supply before starting each transfer.Transfer buffer recommendationsUse only high quality, reagent grade methanol. Contaminated methanol can result in increasedtransfer buffer conductivity, as well as poor transfer of macromolecules. Do not reuse transfer buffers or dilute transfer buffers below recommended levels. Reuse of transfer buffers is not advised, since these buffers have most likely lost their ability to maintain a stable solution pH during transfer. Dilution of transfer buffers below their recommended levels is also not advised, since this will decrease buffering capacity.Voltage limitsDo not increase voltage settings beyond those indicated in Table 3.1. If overnight transfers at low voltages are ineffective for your application, and higher voltages are necessary, transfer times must also be decreased. Failure to do so may result in a potential safety hazard.3.3 Buffer FormulationAll formulas provided below are for a total volume of 1 L of buffer. Approximately 950 ml of buffer are required for the Mini Trans-Blot cell with cooling unit. Ethanol can be used in place of methanol in all buffer formulations.buffersthe electrodes.Note: Some pH electrodes will not perform a proper measurement for the pH of Tris buffers. If the pH of the buffer is off, check to make sure the electrode is designed to work with Tris buffers. If the pH electrode functions properly for Tris buffers and the pH is below 8.0, remake the buffer.25 mM Tris, 192 mM glycine, 20% v/v methanol, pH 8.3 Mix 3.03 g Tris, 14.4 g glycine, and 200 ml of methanol; add distilled deionized water (ddH2O) to 1 L.25 mM Tris, 192 mM glycine, pH 8.3Mix 3.03 g Tris and 14.4 g glycine; add ddH2O to 1 L.48 mM Tris, 39 mM glycine, 20% v/v methanol, pH 9.2 Mix 5.82 g Tris and 2.93 g glycine in ddH2O, add 200 ml methanol.Add to 1 L with ddH2O.48 mM Tris, 39 mM glycine, pH 9.2Mix 5.82 g Tris and 2.93 g glycine.Add ddH2O to 1 L.10 mM NaHCO3, 3 mM NaCO3, 20% methanol, pH 9.9 Mix 0.84 g NaHCO3 and 0.318 g NaCO3 in ddH2O, add 200 ml methanol.Add to 1 L with ddH2O.1.0x TBE (Tris-Borate EDTA), pH 8.390 mM Tris-Borate, 1 mM EDTA5x stock solution54 g Tris base27.5 boric acid20 ml 0.5 M EDTA (pH 8.0)Add 200 ml 5x stock solution to 800 ml ddH2O to make 1x working solution.1x TAE (Tris-Acetate EDTA)40 mM Tris-Acetate, 1 mM EDTA50x stock solution242 g Tris base57.1 ml glacial acetic acid100 ml 0.5 M EDTA (pH 8.0)Add 20 ml 50x stock solution to 980 ml ddH2O to make 1x working solution.Section 4Strategies for Optimizing Electrophoretic Transfer4.1 Optimizing Protein TransferGenerally, quantitative elution of denatured high molecular weight proteins is difficult. The following tactics, alone or in combination, will increase transfer efficiency.Vary gel compositionGradient gels are often more effective than single gel concentrations for elution of a wide range of molecular weight proteins.Lower the total monomer to create a more porous gel. Increase or decrease the percentage of crosslinker. A5.26% C gel will contain the smallest pore size of all gels no matter what the concentration of acrylamide. Decrease in %C will make gels more porous with little loss in resolution.grams bis%C = x 100grams bis + grams acrylamideIncrease transfer timeAn initial control should be performed to determine the time required for complete transfer.18, 25 Times may vary from as little as 30 minutes to as long as overnight. Remember all overnight applications should be performed at 30 volts to minimize heating problems.Increase the powerInitial controls should be performed to evaluate the efficiency of increasing the V/cm as well as its effects on the temperature of transfer. The temperature increase may change buffer resistance and subsequent power delivered, as well as the state of protein denaturation, thus affecting transfer efficiency.Reduce buffer strengthDilution of transfer buffer results in lower current at any given voltage. This will allow the use of higher voltages without excessive heating. However, be aware not to dilute the buffer below its buffering capacity.Vary buffer type and pHMaximize charge-to-mass ratio. It appears that alcohols present in SDS transfer buffer strip SDS from proteins. Basic proteins in Tris, glycine, methanol buffer at pH8.3 may assume a state near isoelectric neutrality and thus transfer poorly. For example, lysozyme exhibits this behavior. Buffers with pH of 9.5–10.0 have shown much better elution and binding characteristics for basic proteins such as lysozyme and histones.41Different buffer types at similar V/cm may yield different efficiencies. Generally, Tris buffers allow more efficient transfer than acetate or phosphate buffers.Add detergentAddition of 0.1% SDS detergent to Tris, glycine, methanol buffer has been reported to increase transfer efficiency.25 SDS, however, increases relative current, power, and heating. Also, temperatures below 10°C may precipitate the SDS so the starting buffer temperature will be higher. SDS may also affect the antigenicity of some proteins. SDS will aid in eluting the proteins from the gel, but it may reduce the binding efficiency of those proteins to the membrane.Eliminate alcohol from the transfer bufferAlcohol in the transfer buffer improves binding of proteins to nitrocellulose only. Elimination of alcohol results in increased transfer efficiency but diminishes binding to nitrocellulose. Transfer efficiency is increased because alcohol causes gel pores to contract resulting in capture of large molecular weight proteins within the gel matrix.Use of PVDF membrane for protein transfers eliminates the alcohol requirement, and constitutes a logical strategy for analysis of high molecular weight or difficult-to-transfer proteins.27, 28 PVDF must be wetted in 100% methanol but may then be used in buffer without methanol.Limited protease treatmentA protocol for protease digestion of protein during transfer has been published.23 Efficient transfer without loss of immunological reactivity was reported.Alter membrane typeBoth nitrocellulose and PVDF can be used for protein transfer.Alter gel systemIf possible, use nondenaturing gradient pore gels for separation of proteins. Isoelectric focusing gels, or native gels, may be considered if separation by molecular weight is not mandatory.Enhance gel-membrane contactFailure of molecules to bind efficiently to the membrane, caused by poor gel-membrane contact, is often confused with inefficient elution. Poor contact is usually due to excess moisture in the gel-membrane interface. Proper technique and the use of a test tube or glass pipet as a “rolling pin” should assure good contact. Proper selection of filter paper spacers will help assure good compression. Gel and membrane equilibration in transfer buffer for 15–20 min prior to transfer will help prevent shrinking of either component during transfer, and will eliminate reactants such as urea or SDS from the gel.4.2 Optimizing DNA and RNA TransferProblems with elution of nucleic acids can be solved by altering the gel percentage. It may be somewhat more difficult to quantitatively transfer large amounts of DNA used in genomic blots. Agarose gels over 6 mm thick are not compatible with the Mini Trans-Blot. The following tactics should be considered for optimizing elution in such transfers.Alter gel compositionLower % total monomer or % crosslinker for polyacrylamide gels.Lower % agarose. This allows better elution of high molecular weight DNA.Alter DNA denaturantsIt has been found that glyoxal denaturation allows more efficient elution of DNA than NaOH. Boiling polyacrylamide gels to denature DNA has also been found to give excellent results.12 Base denaturation often causes polyacrylamide gels to weaken and stick to blotting membranes.Section 5Choice of Blotting Membranes5.1 Protein Blotting MembranesNitrocellulose MembraneNitrocellulose membranes have been used extensivelyfor protein binding and detection.8, 21, 24, 25, 28 They can be easily stained for total protein by a dye stain (Amido Black, Coomassie Blue, Ponceau S, Fast Green FCF, etc.),28 or the more sensitive Colloidal Gold Total Protein Stain, and also allow either RIA, FIA, or EIA.8 Nitrocellulose has a high binding capacity of 80–100 μg/cm2 Nonspecific protein binding sites are easily and rapidly blocked, avoiding subsequent background problems. No pre-activation is required. Low molecular weight proteins (especially <15,000 daltons) may be lost duringpost transfer washes, thus limiting detection sensitivity.20 Smaller pore size nitrocellulose membrane (0.2 μm),has been shown to be effective in eliminating this loss.30 Large proteins (>100,000 daltons) denatured by SDS may transfer poorly due to the addition of alcohol to the transfer buffer. Alcohol increases binding of SDS-proteins to nitrocellulose, but decreases pore sizes in the gel. Elimination of alcohol from SDS-protein transfers results in considerably diminished binding. Adding SDS (up to 0.1%) to the transfer buffer increases the transfer efficiencyof proteins, but reduces the amount of binding to the membrane.18 Also, SDS increases the conductivity of the buffer and the heat generated during transfer.PVDF MembranePolyvinylidene difluoride (PVDF) membrane is an ideal support for amino-terminal sequencing, amino acid analysis and immunoassays of blotted proteins. PVDF retains proteins under extreme conditions of exposure to acidic or basic conditions, and in the presence of organic solvents.Greater retention during sequencing manipulations enhances the likelihood of obtaining information from rare, low abundance proteins, by increased initial coupling and higher repetitive yields. In addition, PVDF membrane exhibits better binding efficiency of blotted material in the presence of SDS in the transfer buffer. PVDF must firstbe wetted in 100% MeOH but can then be used in buffer, which does not contain MeOH.5.2 DNA and RNA Blotting MembranesZeta-Probe® Nylon MembraneNitrocellulose is not a suitable medium for electrophoretic transfer of nucleic acids, as high concentrations of salt (>10x SSC) are required for efficient binding.13 Molecules ≤500 bp are not bound at all, even at high salt. Low resistance results when an electric current is passed through a solution of high salt. This causes potentially damaging high currents (and power) even at very low voltages. Since V/cm is the eluting force, inefficient transfer occurs under conditions required for proper binding. Zeta-Probe membrane allows efficient binding of all sizes of single stranded DNA and RNA in the presence of low ionic strength buffers.13 Zeta-Probe membraneis an ideal alternative to nitrocellulose for the transfer of nucleic acids. Binding is more stable through post transfer washes, and reprobing may be performed as many as 10 times.A variety of blotting membranes is available for immunoblotting, each with particular advantages depending on the needs of the experiment. The physical properties and performance characteristics of a membrane should be evaluated when selecting the appropriate transfer conditions.Table 5.1 Guide to Protein Blotting MembranesMembrane Pore Size BindingCapacity(μg/cm 2)Notes Nitrocellulose0.45 μm 0.2 μm 80–100General purpose protein blotting membrane.SupportedNitrocellulose 0.45 μm 0.2 μm 80–100Pure nitrocellulose cast on an inert synthetic support; increasedstrength for easier handling andfor reprobing.PVDF 0.2 μm 170–200High mechanical strength andchemical stability, used for proteinsequencing and western blotting;enhanced binding in the presenceof SDS. Must be wet in alcoholbefore equilibration in buffer.Nylon 0.2 μm 170Recommended for nucleic acids.Note : Nucleic acids cannot be transferred to nitrocellulose by electrophoretic blotting. Use Zeta-Probe membrane.。


高温高压封隔器的研制及应用孙德启(上海优强石油科技有限公司,上海201806)摘要:针对高温、高压、高钢级套管、高比重泥浆等的井下苛刻作业环境导致完井工具稳定性大幅度降低,研制了高温高压H S I I封隔器。
该封隔器在设计上充分考虑了高温密封件的耐压性能以及卡瓦对硬度较高套管的锚定。
尤其采用了高强度卡瓦和软金属胶筒护肩设计,确保封隔器在恶劣井下环境中长期工作的稳定可靠。
现场应用结果表明,该封隔器适用于高温、高压等复杂井况下的完井作业,成功率高。
关键词:封隔器;高温高压;软金属护肩;坐封中图分类号:T E931文献标识码:A文章编号:1001 -196X(2018)04 -0041 -05Development and application of hij=h temperature and hij=h pressure packerSUNDe-qi(Shanghai Extrong Oilfield Technology C o.,L t d.,Shanghai 201806, C h i n a)Abstract: T h e harsh working environment such as H T H P,high-grade casing a n d high specific gravity m u d,m a k e s the stability o f completion tool greatly reduced. T h e high temperature a n d high pressure! H T H P) HSIIpacker has b e e n developed. It is designed with considerations to pressure resistance of higha n d the slip’s a nchorage to the high hardness casing. In particular,b y using high strength slips a n d soft metalshoulder protections,i t could ensure that packers w o r k stably a n d reliably in b a d down-hole environment tor along time. T h e field application results s h o w that tlie packer is suitable for completion operation un d e rcated well conditions s uch as H T H P,a n d improve the success rate.Keywords:p a c k e r;high temperature a n d high pressure;soft metal shoulder protection;setting〇前言国内油气资源经过几十年的不断勘探开发,新发现油气资源的埋藏深度越来越深(深层油气 资源埋藏深度在5 000 m以下),开采难度越来 越大,对完井工具的要求也越来越高,对封隔器 的耐温、耐压、受力均提出了较高的要求[1-4]。

代容春,林荣华,何文锦,等. 湖泊红球藻等离子诱变及其高产虾青素藻株培养条件的优化[J]. 食品工业科技,2023,44(23):213−220. doi: 10.13386/j.issn1002-0306.2023030120DAI Rongchun, LIN Ronghua, HE Wenjin, et al. Plasma Mutagenesis of Haematococcus lacustris and Optimization of Culture Conditions for High-yield Astaxanthin Algae Strains[J]. Science and Technology of Food Industry, 2023, 44(23): 213−220. (in Chinese with English abstract). doi: 10.13386/j.issn1002-0306.2023030120· 工艺技术 ·湖泊红球藻等离子诱变及其高产虾青素藻株培养条件的优化代容春1,2, *,林荣华1,2,何文锦1,2,薛 婷1,2,陈建楠1,2,陈 菁1,2,孙化淼1,2(1.福建师范大学生命科学学院,福建福州 350117;2.福建师范大学南方海洋研究院,福建福州 350117)摘 要:为进一步提高湖泊红球藻(Haematococcus lacustris )的工业利用价值,本研究使用常压室温等离子体(atmospheric and room temperature plasma ,ARTP )诱变仪对湖泊红球藻进行等离子诱变。
以藻细胞致死率为指标确定等离子诱变的适宜输入功率和诱变时间。
诱变之后通过固体平板培养初筛和液体培养复筛获得高产虾青素的突变藻株。
再以藻细胞密度为指标采用单因素实验及正交试验对高产藻株的营养生长阶段的培养条件进行优化,并筛选虾青素诱导阶段适宜虾青素积累的高光照条件。

离子推进器上限速度英文回答:The maximum speed of an ion thruster is determined by several factors, including the design and efficiency of the thruster, the available power source, and the specific propellant being used. In general, ion thrusters can achieve speeds of up to 50,000 meters per second (m/s) or more.The speed of an ion thruster is mainly limited by the exhaust velocity of the propellant ions. The exhaust velocity is determined by the energy imparted to the ions by the thruster's electric field. The higher the exhaust velocity, the faster the thruster can accelerate the ions and the higher the maximum speed it can achieve.One example of a high-speed ion thruster is the Hall effect thruster. This type of thruster uses a magneticfield to confine the plasma and increase the exhaustvelocity of the ions. Hall effect thrusters can achieve speeds of up to 30,000 m/s or more, making them suitablefor long-duration space missions.Another example is the gridded ion thruster, which uses a series of charged grids to accelerate and expel ions. Gridded ion thrusters can achieve speeds of up to 50,000m/s or more, but they are typically less efficient than Hall effect thrusters.It's important to note that while ion thrusters can achieve high speeds, they do so at a relatively low thrust. This means that it takes a longer time for an ion thruster to reach its maximum speed compared to traditional chemical rockets. However, ion thrusters are highly efficient and can operate for extended periods of time, making them ideal for long-duration space missions where speed is not the primary concern.中文回答:离子推进器的最高速度由几个因素决定,包括推进器的设计和效率、可用的功率源以及使用的特定推进剂。
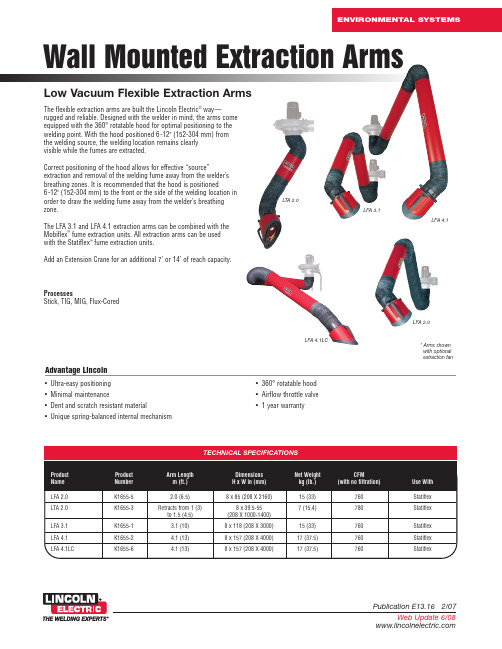
Wall Mounted Extraction ArmsLow Vacuum Flexible Extraction ArmsAdvantage Lincoln•Ultra-easy positioning •Minimal maintenance•Dent and scratch resistant material •Unique spring-balanced internal mechanism•360°rotatable hood •Airflow throttle valve •1year warrantyThe flexible extraction arms are built the Lincoln Electric ®way—rugged and reliable.Designed with the welder in mind,the arms come equipped with the 360°rotatable hood for optimal positioning to the welding point.With the hood positioned 6-12"(152-304mm)from the welding source,the welding location remains clearly visible while the fumes are extracted.Correct positioning of the hood allows for effective “source”extraction and removal of the welding fume away from the welder’s breathing zones.It is recommendedthat the hood is positioned6-12"(152-304mm)to the front or the side of the welding location in order to draw the welding fume away from the welder’s breathing zone.The LFA 3.1and LFA 4.1extraction arms can be combined with the Mobiflex ™fume extraction units.All extraction arms can be used with the Statiflex ™fume extraction units.Add an Extension Crane for an additional 7’or 14’of reach capacity.ProcessesStick,TIG,MIG,Flux-CoredLTA 2.0LFA 4.1LCLFA 4.1LFA 2.0LFA 3.1*Arms shown with optional extraction fanPERFORMANCEThe extraction arms are made of a solid,lightweight aluminum internal skeleton and a 203mm (8")diameter dent and scratch resistant outer plastic shell.Incorporated into the arm is a throttle valve that can be set to a low,partial,full or no airflow setting at the hood opening.The arms also feature an extraction focus spoiler,which directs the air into the hood.Lamp Kits or Lamp Kits with Arc Sensors are available as recommended options.With the use of an arc sensor,the automatic start/stop utilizes a 20second auto stop to help conserve energy.All this combined with easy positioning provides a highly effective system.The arm’s balance system provides durability,stability and easy positioning.When the arm is raised,it is free to move in any direction.When the arm is positioned,it is fixed into place.This makes re-positioning the arms very light and easy.Low vacuum extraction systems utilize high volume/low velocity air movement to enable the arm to be positioned at a distance of 6-12"(152-304mm)from the welding source.This allows a larger work zone with less repositioning of the arm and increased user visibility to the weld location.Source extraction is a term used to describe the method of extracting or capturing and filtering the welding fume at ornear the welding arc or source.If applied correctly,this process effectively draws the welding fume away from the operator’s breathing zone as well as greatly reduce the residual fume within the welding facility.Statiflex ™200-M Dual ArmLTA 2.0LFA2.0360°Rotatable HoodGENERAL OPTIONSSF2400FanThe 1HP ,115/1/60fan hasself-cleaning concave blades and provides a maximum open air flow of 1200CFM with extraction arm.Requires a mounting bracket.Order K1656-1SF2400Wall Mounting Bracket Use K1657-1wall mountingbracket for use with LFA 3.1,4.1or e K1657-2bracket with LFA 2.0and LTA 2.0telescopic extraction arm or EC 2and EC 4extension cranes.OrderK1657-1for LFA 3.1and 4.1K1657-2for LTA 2.0,EC 2,and EC 4SF4200FanThe 2HP ,230/3/60fan hasself-cleaning concave blades and provides a maximum open airflow of 1750CFM with extraction arm.Includes wall mounting bracket.Order K1656-4Lamp Kit with Arc SensorMounted in the lamp housing,the arc sensor turns the fan on when sensing the arc flash,and turns off 20seconds after the arc ends.Hood mounted switches turn the unit on and off independently of the arc sensor.Statiflex ™Kit includes:lamp housing with arc sensor,control box,interconnect wire,hood mounted lamp/fan switch,and instruction manual.OrderK1669-4SF2400Fan K1669-10SF4200Fan230V Conversion Kit for SF2400FanProvides 230V operation of the SF2400Fan when using Lamp Kit or Lamp Kit with Arc Sensor.Order K1750-1SF2400Starter Overload Switch for 115VProvides simple on/off operation when a Lamp Kit or Lamp Kit with Arc Sensor is NOT used.This overload switch retrofits theextraction unit for 115V operation of SF2400Fan.Order K1494-2SF2400/SF4200Starter Overload Switch for 230VProvides simple on/off operation when a Lamp Kit or Lamp Kit with Arc Sensor is NOT used.This overload switch retrofits theextraction unit for 230V operation of fan.OrderK1494-3SF2400Fan K1494-10SF4200FanEC 2(7ft.)and EC 4(14ft.)Extension CranesFor large work areas.Extend your reach 2meters (7ft.)or 4meters (14ft.).A side-to-side hinge has a unique spring controlled lock to hold the arm in place for steady positioning.Each crane includes support boom,mounting plate,8"tubing,flexible hose,and mounting brackets.OrderK1671-1for EC 2K1671-2for EC 4Feeder Hanger ArmUse with extension cranes to slide mount a wire feeder on the underside of the crane.OrderK1672-1Statilflex KitShownC U S T O M E R A S S I S T A N C E P O L I C YThe business of The Lincoln Electric Company is manufacturing and selling high quality welding equipment,consumables,and cutting equipment.Our challenge is to meet the needs of our customers and to exceed their expectations.On occasion,purchasers may ask Lincoln Electric for advice or information about their use of our products.We respond to our customers based on the best information in our possession at that time. Lincoln Electric is not in a position to warrant or guarantee such advice,and assumes no liability,with respect to such information or advice.We expressly disclaim any warranty of any kind,including any warranty of fitness for any customer’s particular purpose,with respect to such information or advice.As a matter of practical consideration,we also cannot assume any responsibility for updating or correcting any such information or advice once it has been given,nor does the provision of information or advice create,expand or alter any warranty with respect to the sale of our products.Lincoln Electric is a responsive manufacturer,but the selection and use of specific products sold by Lincoln Electric is solely within the control of,and remains the sole responsibility of the customer.Many variables beyond the control of Lincoln Electric affect the results obtained in applying these types of fabrication methods and service requirements.Subject to Change–This information is accurate to the best of our knowledge at the time of printing.Please refer to for any updated information.。


如何回复审稿人意见:意见1:所有问题必须逐条回答。
2.尽量满足意见中需要补充的实验。
3.满足不了的也不要回避,说明不能做的合理理由。
4.审稿人推荐的文献一定要引用,并讨论透彻。
5. 老师说的4点,确实很有道理。
不过审稿人提出要补充的实验,如果不是非做不可的,还是可以进行解释。
我也为国外的杂志审过稿,有时审稿人即使想接受你的文章,总还要提出一些不足之处,如果文章没有那些不足之处,也许文章就会投给更高IF的杂志了。
所以,如果你真的不想补充实验或者补充很困难,可以合理的解释,一般没问题的。
国外杂志要求补充实验的,我均以解释而过关,原因见少帖)。
还因为:很少杂志编辑把你的修改稿再寄给当初审稿人的,除非审稿人特别请求。
编辑不一定懂你的东西,他只是看到你认真修改,回答疑问了,也就接受了(当然高档杂志可能不是这样,我的经验只限定一般杂志(影响因子1-5)。
我常用的回复格式,呵呵。
Dear reviewer:I am very grateful to your comments for the manuscript. According with your advice, we amended the relevant part in manuscript. Some of your questions wereanswered below.引用审稿人推荐的文献的确是很重要的,要想办法和自己的文章有机地结合起来。
至于实验大部分都可以不用补做,关键是你要让审稿人明白你的文章的重点是什么,这个实验对你要强调的重点内容不是很必要,或者你现在所用的方法已经可以达到目的就行了。
最后要注意,审稿人也会犯错误,不仅仅是笔误也有专业知识上的错误,因为编辑找的审稿人未必是你这个领域的专家。
只要自己是正确的就要坚持。
在回复中委婉地表达一下你的意见,不过要注意商讨语气哦!我的回复,请老外帮忙修改了Dear Editor:Thank you for your kind letter of “......” on November **, 2005. We revised the manuscript in accordance with the reviewers’comments, and carefully proof-read the manuscript to minimize typographical, grammatical, and bibliographical errors.Here below is our description on revision according to the reviewers’ comments.Part A (Reviewer 1)1. The reviewer’s comment: ......The authors’ Answer: .....2. The reviewer’s comment: ......The authors’ Answer: .....Part B (Reviewer 2)The authors’ Answer:Many grammatical or typographical errors have been revised.All the lines and pages indicated above are in the revised manuscript.Thank you and all the reviewers for the kind advice. Sincerely yours,具体例子1:这是我的一篇修稿回复,杂志是JBMR-A,影响因子3.652,已发表,供参考!Reply to the comments on JBMR-A-05-0172Comment:Reference #10 is missing from the Introduction but used much later in the manuscript. Should these be in order used in manuscript?Reply:The missing reference has been added into the revisedmanuscript.Comment (continued):What is the sample size for all tests performed?Reply:The sample size for drug release and PCL degradation tests was 3.0×3.0 cm2, with a thickness of about 0.1mm and a weight of about 40mg. This dada have been added into the revised manuscript.Comment (continued):Figure 7. There is no scientific evidence presented in the TEM figure to convince this reviewer of sub-jets. This statement on Page 9 cannot be made without clear evidence during the jet formation/separation. Figure 7 is just a large fiber and small fiber fused together, no other conclusion than this can be made.Reply:Necessary change in the statements has been made in the revised manuscript as well as in the referredfigure accordingly.Comment (continued):Table 3: Need standard deviation for all values reported not just for a select few.. Equation after Table 3 not necessary. Just reference method used.Reply:Done accordingly.Comment (continued):Page 11: "faster weight loss" What was the sample size? Where is the statistical analysis of this data? This reviewer does not see a significant difference in any of the data presented, thus weight loss would be considered equivalent.Reply:Although not too much difference was seen, the conclusion that “the GS/PCL membrane exhibited a relatively faster weight loss compared with the RT/PCL membrane” was indeed applicable through “one-way analysis of variance (ANOVA)” analysis. Following the reviewer’s comment, a new sub-section has been added to the manuscript to address the statistical analysis for the data.Comment (continued):Page 12: What is the sample size for release data? Looks like results based on a sample size of one? Need stand deviations on the data presented in Figure 11.Why wasn't release performed and compared for all electrospun conditions investigated otherwise?Reply:Three repeated tests were performed for each set of measurements and the resulting data were averaged. As stated in the revised manuscript, each sample had a square area of 3cm2 with a slightly different thickness. 3Standard deviations have been added to the data shown in Fig. 11.The present manuscript aimed to show that medicaldrugs can be encapsulated in ultrafine fibers through a co-axial electrospinning process. The drug release data intended to show that the encapsulation was successful. We did not consider any specific application in this preliminary paper, and in fact the two drugs were just chosen as model illustration. As such, there seemed not necessary to perform release experiments for all of the membranes electrospun with different conditions (i.e. the core concentrations)Comment (continued):Table 3: Yang's or Young's Modulus (page 10 says Young's).Reply:Corrected accordingly.Comment (continued):Figure 11: What is the % release, not just concentration. Why just this small sample of release data? Where is the release data for the other conditions?Reply:Unfortunately, we did not measure the amount of the shell material in obtaining the composite nanofibers. Namely, the flow rate of the shell solution during the electrospinning was not accurately controlled using an injecting pump. Hence the % release was not applicable.Please refer to the previous reply related to Page 12 and Figure 11 for the remaining comments.We acknowledge the reviewer’s comments and suggestions very much, which are valuable in improving the quality of our manuscript.具体例子2:Major comments:1. The authors need to strengthen their results by including MMPsecretion, and tran-matrigel migration by a positive controlprogenitor cell population i.e. enriched human CD34 cellsobtained from mobilized PBL, since this is a moreclinicallyrelevant source of CD34 cells which has also been shown tosecrete both MMP-9 and MMP-2 (ref. 11). CD34 enriched cellsfrom steady state peripheral blood which also secrete MMPs arealso of interest.2. In fig 1C please specify which cell line represents MMP-negative cells. This needs to be clarified, as well as abetter explanation of the method of the protocol.3. The ELISA results are represented as "fold increase" comparedto control. Instead, we suggest that standards should be used andresults should be presented as absolute concentrations and onlythen can these results be compared to those of the zymography.4. When discussing the results, the authors should distinguishclearly between spontaneous migration vs chemotactic migration.Furthermore, the high spontaneous migration obtained with cordblood CD34 cells should be compared to mobilized PBL CD34enriched cells and discussed.5. The authors claim that the clonogenic assay was performed todetermine the optimum concentration for inhibition of MMPactivity by phenanthroline and anti MMP-9 mAb, however theyshould clarify that this assay can only determine the toxicity ofthe inhibitors and not their optimal inhibitory concentrations.Minor comments:1. There are many spelling and syntax errors, especially in theresults and discussion, which need correction.a. Of special importance, is the percent inhibition of migration,which is described as percent of migration. i.e. pg 7:"Migrationof CB CD34 was reduced to 73.3%?" Instead should read "Migration of CB CD34 was reduced by 73.3%?"b. The degree symbol needs to be added to the numbers inMaterials and methods.2. It would be preferable to combine figure 1A and B, in order toconfirm the reliability of fig. 1B by a positive control(HT1080).Answer to referee 1 comment:1. Mobilized peripheral blood is a more clinical source of CD34+ cells, so it is necessary to compare the MMP-9 secretion and trans-migration ability of CB CD34+ cells with that of mobilized PB CD34+ cells. However, we couldn't obtain enough mobilized PB toseparate PB CD34+ cells and determine the MMP-9 secretion and migration ability, so we couldn’t complement the study on PB CD34+ cells in this paper. Results obtained by Janowska-Wieczorek et al found that mobilized CD34+ cells in peripheral blood express MMP-9. Furthermore, Domenech’s study showed that MMP-9 secretion is involved in G-CSF induced HPC mobilization. Their conclusions have been added in the discussion. In our present study, our central conclusion from our data is that freshly isolated CD34+ stem/progenitor cells obtained from CB produce MMP-9.2. MMP-9 negative cell used in fig 1C was Jurkat cell. In zymographic analysis, MMP-9 was not detected in the medium conditioned by Jurkat cell. To exclude that the contaminating cells may play a role in the observed MMP-9 production, we screened the media conditioned by different proportion of CB mononuclear cells with MMP-9 negative cells by zymography. This result may be confusion. Actually, only by detecting the medium conditioned by 2X105 CB mononuclear cells(MNC)/ml (since the purities of CD34+ cell are more than 90%), it could exclude the MNC role. In the revised manuscript, we only detected MMP-9 activity and antigen level in the medium conditioned by 2X105 CB mononuclear cells (MNC)/ml. There is no MMP-9 secretion be detected in the medium conditioned by 2X105 CB MNC/ml. It excluded the possibility that the MMP-9 activity in CB CD34+ cells conditioned medium is due to the contamination by MNC.3.In this revised paper, we have detected the MMP-9 antigen levels by using commercial specific ELISA kits (R&D System, sensitivity, 0.156ng/ml). Recombinant MMP-9 from R&D System was used as a standard. The results are expressed in the absolute concentration. The absolute concentration result has been added in the paper. As shown in Fig2, MMP-9 levels were detectable in both CB CD34+ cell conditioned medium and BM CD34+ cell conditioned medium. However, MMP-9 level was significantly higher in CB CD34+ cell conditioned medium than in BM CD34+ cell conditioned medium (0.406±0.133ng/ml versus0.195±0.023ng/ml). Although gelatinolytic activity was not detected in media conditioned by CD34+ cells from BM, sensitivity of ELISA favors the detection of MMP-9 antigen in the BM CD34+.4. In our study, to establish the direct link between MMP-9 and CB CD34+ cells migration, we only determined the role of MMP-9 in spontaneous migration of CB CD34+ cells, but not in chemotactic migration. Actually, regulation of hematopoietic stem cell migration, homing and anchorage of repopulation cells to the bone marrow involves a complex interplay between adhesion molecules, chemokines, cytokines and proteolytic enzymes. Results obtained by the groups of Voermans reveal that not only the spontaneous migration but also the SDF-1 induced migration of CB CD34+ cells is greatly increased in comparison to CD34+ cells from BM and peripheral blood.5. CD34+ cells we obtained in each cord blood sample were very limited. It is not enough to screen the inhibitors concentrations to select the optimalinhibitory concentrations. In the blocking experiments, based on the concentrations used by others and the manufacturer's recommendation, we then determined the inhibitors concentrations by excluding the toxicity of the inhibitors in that concentration, which was determined by clonogenic assay.Minor comments:1.The spelling and syntax errors have been checked and corrected.2.Since the results in figure 1A and B were obtained from two separated and parallel experiments, it is not fitness to combine two figures.下面把我平时总结的一些答复审稿人的策略和写回复信的格式和技巧跟大家交流一下。

qScript™ One-Step SYBR® Green qRT-PCR Kit, ROX™Cat No. 95088-050Size: 50 x 50-µL reactions Store at -25ºC to - 15°Cprotected from light 95088-200200 x 50-µL reactionsDescriptionThe qScript One-Step SYBR Green qRT-PCR Kit, ROX is a convenient and highly sensitive solution for reverse transcription quantitative PCR (RT-qPCR) of RNA templates using SYBR Green I dye detection and gene-specific primers on Applied Biosystems 7000, 7300, 7700, 7900HT StepOne™, or StepOnePlus™ real-time PCR systems. cDNA synthesis and PCR amplification are carried out in the same tube without opening between procedures. The system has been optimized to deliver maximum RT-qPCR efficiency, sensitivity, and specificity. The proprietary reaction buffer has been specifically formulated to maximize activities of both reverse transcriptase and Taq DNA polymerase while minimizing the potential for primer-dimer and other non-specific PCR artifacts. The kit is compatible with both fast and standard qPCR cycling protocols. Highly specific amplification is essential for successful RT-qPCR with SYBR Green I technology, since this dye binds to any dsDNA generated during amplification. AccuStart™ Taq DNA polymerase contains monoclonal antibodies that bind to the polymerase and keep it inactive prior to the initial PCR denaturation step. Upon heat activation at 95ºC, the antibodies denature irreversibly, releasing fully active, unmodified Taq DNA polymerase. Instrument CompatibilityDifferent real-time PCR systems employ different strategies for the normalization of fluorescent signals and correction of well-to-well optical variations. It is critical to match the appropriate qPCR reagent and internal reference dye to your specific instrument. The qScript Custom One-Step SYBR Green qRT-PCR Kit, ROX provides seamless integration on the Applied Biosystems 7000, 7300, 7700, 7900, 7900HT, StepOne™, or StepOnePlus™. Please visit our web site at to find the optimal kit for your instrument platform.ComponentsReagent Description 95088-050 95088-200qScript One-Step Reverse Transcriptase Optimized 50X formulation of recombinant MMLV reverse transcriptase for one-step RT-PCR.1 x 50 µL 1 x 200 µLOne-Step SYBR Green Master Mix, ROX (2X) 2X reaction buffer containing dNTPs, magnesium chloride, AccuStart Taq DNApolymerase, stabilizers, ROX reference dye and SYBR Green I dye1 x 1.25 mL 4 x 1.25 mLNuclease-free water 1 x 1.5 mL 4 x 1.5 mLStorage and StabilityStore components in a constant temperature freezer at -25°C to -15°C protected from light upon receipt.For lot specific expiry date, refer to package label, Certificate of Analysis or Product Specification Form.Guidelines for One-Step SYBR Green qRT-PCR▪Primer design is critical for successful one-step RT-qPCR with SYBR Green. The use of software tools for PCR primer design and RNA secondary structure analysis can aide in the design of specific and efficient primers for one-step RT-qPCR. Primers should be designed according to standard qPCR guidelines with a length of 18 - 25 nucleotides and a GC content of 40-65%. Avoid internal secondary structure, and complementation at 3’ ends within each primer and primer pair. 3’-end terminal stability should be kept low to maximize primer specificity (3’-pentamer ΔGº > -8.0 kcal/mol or have no more than 2 to 3 Cs or Gs in the last 5 bases).▪Regions of RNA secondary structure should be avoided as this can interfere with annealing of the reverse primer for cDNA synthesis and/or impede procession of the reverse transcriptase. Programs for RNA structure prediction, such as the mfold web server (/), are useful for selecting regions of relaxed RNA structure for qRT-PCR primer design.▪Ideally, primer Tm should be between 58 and 60ºC for a typical 2-step qPCR cycling protocol. Estimation of primer Tm varies widely with different methods and analysis parameters. We recommend using a program that calculates Tm based on nearest-neighbor thermodynamic models at 50 mM monovalent salt and 50 nM primer concentration. Primers with melting temperatures outside of this range may require optimization of PCR cycling conditions.▪PCR product size should be between 70- 200 bp. Ideally, the amplified sequence should span intronic sequence to minimize the potential to amplify genomic DNA sequence. Design primers to anneal to exons that bracket intronic sequence or within exon / exon boundaries of the s pecific mRNA. NCBI’s Primer-BLAST program (/tools/primer-blast/index.cgi?LINK_LOC=BlastHomeAd) can facilitate the design of RNA-specific primer sets.Control reactions that lack reverse transcriptase (minus RT) should always be included to verify that amplification signal is due to the presence of RNA target and not genomic DNA.▪ A final concentration of 200 nM each primer is recommended as a general starting point. Optimal results may require titration of primer concentration between 100 and 500 nM. PCR efficiency is often improved with higher primer concentration (300 to 500 nM). In some cases, higher concentration of the reverse primer alone may improve RT-PCR efficiency without compromising specificity. We highly recommend including a post PCR dissociation analysis step (melt curve) to distinguish specific from non-specific amplification product(s) (i.e. primer-dimer).▪Thaw all components, except the qScript One-Step RT, at room temperature. Mix by gently vortexing, then centrifuge to collect contents to the bottom of the tube before using. Place all components on ice after thawing.▪To maximize assay specificity and sensitivity reactions should be assembled on ice and kept cold until placed in your real-time PCR system. Centrifugation steps should be carried out in a refrigerated centrifuge. AccuStart Taq DNA polymerase is inactive prior to high temperature activation; however, reverse transcriptases are active at lower temperatures and can use single strand DNA as a template.Guidelines for One-Step SYBR Green qRT-PCR continued:▪ First-strand synthesis can be carried out between 42°C and 52°C. Optimal results are generally obtained with a 5-minute incubation at 50°C. We recommend a 2-5 minute incubation at 95°C to fully inactivate the RT prior to PCR cycling.▪Preparation of a reaction cocktail is recommended to reduce pipetting errors and maximize assay precision. Assemble the reaction cocktail with all required components except RNA template and dispense equal aliquots into each reaction tube. Add RNA to each reaction as the final step. Addition of sample as5 to 10-µL volumes will improve assay precision.▪Suggested input quantities of template are: 1 pg to 100 ng total RNA; 10 fg to 100 ng poly A(+) RNA; 10 to 1x108 copies viral RNA.▪After sealing each reaction, vortex gently to mix contents. Centrifuge briefly to collect components at the bottom of the reaction tube.Reaction AssemblyComponent Volume for 50-μL rxn. Final ConcentrationOne-Step SYBR Green Master Mix, ROX (2X) 25 µL 1XForward primer Variable 200 – 300 nMReverse primer Variable 200 – 300 nMNuclease-free water VariableRNA template 5 – 10 µL VariableqScript One-Step RT * 1 µL 1XFinal Volume (μL) 50 µLNote: Reaction volume can be scaled from 5 to 50 µL depending on the reaction plate (i.e. 384-well vs. 96-well) and qPCR system. Scale all component volumes proportionally. * Omit addition of qScript One-Step RT in minus RT control reactions.Reaction ProtocolIncubate the complete reaction mix in a real-time thermal detection system as follows:Fast qPCR Cycling Standard qPCR Cycling 3-Step PCR Cycling cDNA Synthesis 50°C, 5 min 48 – 50°C, 10 min 48 – 50°C, 10 minTaq Activation 95°C, 2 min 95°C, 5 min 95°C, 5 minPCR cycling (30 - 45 cycles) 95°C, 3s 95°C, 10s 95°C, 10s60°C, 30s (data collection) 60°C, 30s (data collection) 55 – 65°C, 20s68 – 72°C, 30 to 60s (data collection)Melt Curve (dissociation stage): See instrument instructions See instrument instructions See instrument instructionsOptimal cycling conditions will vary for different primer sets. A 3-step cycling protocol may improve assay specificity with some primer sets.Quality ControlKit components are free of contaminating DNase and RNase. The qScript One-Step SYBR Green qRT-PCR Kit, ROX is functionally tested in RT-qPCR. Kinetic analysis must demonstrate linear resolution over six orders of dynamic range (r2 > 0.995) and an RT-PCR efficiency > 90%Limited Label LicensesUse of this product signifies the agreement of any purchaser or user of the product to the following terms:1.The product may be used solely in accordance with the protocols provided with the product and this manual and for use with components contained in the kitonly. QIAGEN Beverly, Inc. grants no license under any of its intellectual property to use or incorporate the enclosed components of this kit with any components not included within this kit except as described in the protocols provided with the product, this manual, and additional protocols available at . Some of these additional protocols have been provided by Quantabio product users. These protocols have not been thoroughly tested or optimized by QIAGEN Beverly, Inc.. QIAGEN Beverly, Inc. neither guarantees them nor warrants that they do not infringe the rights of third-parties.2.Other than expressly stated licenses, QIAGEN Beverly, Inc. makes no warranty that this kit and/or its use(s) do not infringe the rights of third-parties.3.This kit and its components are licensed for one-time use and may not be reused, refurbished, or resold.4.QIAGEN Beverly, Inc. specifically disclaims any other licenses, expressed or implied other than those expressly stated.5.The purchaser and user of the kit agree not to take or permit anyone else to take any steps that could lead to or facilitate any acts prohibited above. QIAGEN Beverly,Inc. may enforce the prohibitions of this Limited License Agreement in any Court, and shall recover all its investigative and Court costs, including attorney fees, in any action to enforce this Limited License Agreement or any of its intellectual property rights relating to the kit and/or its components.©2018 QIAGEN Beverly Inc. 100 Cummings Center Suite 407J Beverly, MA 01915Quantabio brand products are manufactured by QIAGEN, Beverly Inc.Intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.qScript and AccuStart are trademarks of QIAGEN Beverly, Inc. SYBR is a registered trademark of Molecular Probes, Inc. StepOne, StepOnePlus, and ROX are trademarks of Life Technologies Corporation.。
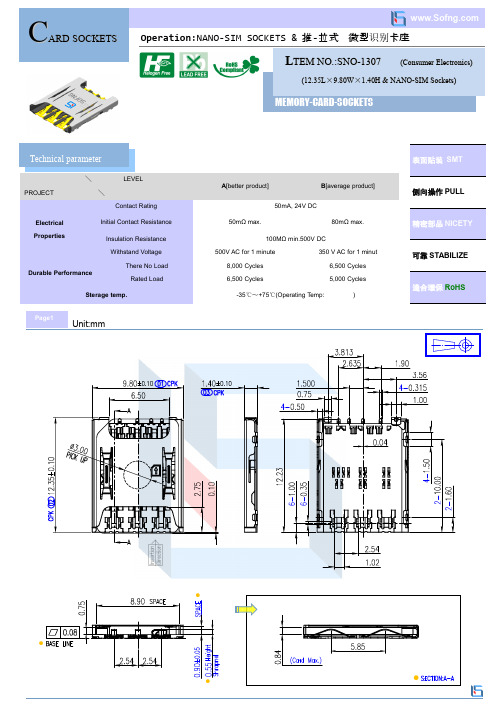

3B SCIENTIFIC ® PHYSICS1Instruction sheet05/18 TLE/UD1 Connecting cable, miniDIN2 Push-button for distant zone3 Light transmitter and lightreceiver4Push-button for near zoneThe laser reflection sensor meets the safety regulations for Class 2 lasers. It emits light in the visible region of the spectrum (400 – 700 nm). The radiation power is less than 1 mW. Provided that the instrument is used in accord-ance with the instructions, safe operation is ensured.In schools and other places of education or training, the instrument must only be used under the supervision of a trained and respon-sible person.Do not look into the light beam - that is not necessary when using the sensor.∙ If the housing of the instrument showsvisible signs of damage, it must be taken out of use immediately. ∙ Do not use any optical equipment that nar-rows the light beam. ∙Do not open the instrument housing.The instrument emits a laser beam at 630 nm wavelength and measures the reflected light. Two sensitivity ranges (near zone and distant zone) are provided, to adjust the instrument parameters for different operating conditions. The near-zone setting adjusts the instrument pa-rameters for operating distances of 5 to 50 mm. The experiment should be chosen so that there are widely different reflectivities (e.g. matt black markings on a white or diffusely reflecting background). With reflecting foils or microprism mirrors, a larger fraction of the light is reflected in the incident direction, and one can then work with a beam box up to 5 m long without special adjustments to the mirror. In such cases one should use the distant-zone setting.By connecting the instrument to other external technical aids, one can measure physical quantities related to the motions of bodies, such as rate of rotation, angle of rotation, an-3B Scientific GmbH ▪ Rudorffweg 8 ▪ 21031 Hamburg ▪ Germany ▪ Subject to technical amendments © Copyright 2018 3B Scientific GmbHgular acceleration, distance travelled, velocity and linear acceleration.The sensor is compatible with the VinciLab (1021477), the €Lab (1021478) and the digital counter (1001032 or 1001033). To perform experimen ts using the VinciLab and the €Lab the connection cable MiniDIN8-BT (1021688) is additionally required. Through the connec-tion box (1009954 or 1009955) it is possible to link the sensor to any of the other instrument technologies via 4 mm sockets.A magnet in the base of the sensor provides a convenient means of locating it firmly.1 Laser reflection sensor 1 MiniDIN connecting cable 1 Piece of reflecting foilLight source: Laser module, 630 nm wavelength Beam divergence: approx. 1 mrad Max. light power: 500 µW Laser class: II Dimensions: 40 x 25 x 90 mm 3 Mass: approx. 0.05 kgNo special maintenance procedures are nec-essary. ∙ Do not use any aggressive cleaning agents or solvents to clean the equipment. ∙ Use a soft, damp cloth for cleaning.∙ The packaging should be disposed of at local recycling points. ∙Should you need to dispose of the equip-ment itself, never throw it away in normal domestic waste. If be-ing usedin private households it can be disposed of at the local public waste disposal authority.∙ Comply with the applicable regulations for the disposal of electrical equipment.。

Shipping: On Dry/Blue ice Catalog numbers:BIO -25045: 200 x 50 μL reactions 4 x 1.25 mLBatch No.: See vial BIO -25046: 1000 x 50 μL reactions 20 x 1.25 mL Concentration: 2xStore at –20 °CDescriptionMyTaq ™ HS Mix is a ready -to -use 2x mix for fast, highly -specific, hot -start PCR. MyTaq HS Mix is powered by antibody mediated hot -start and does not possess polymerase activity during the reaction set -up, thus reducing non -specific amplification. The advanced formulation of MyTaq HS Mix allows fast cycling conditions to be used, greatly reducing the reaction time without compromising PCR specificity and yield. Thanks to its speed and high specificity MyTaq HS Mix is also highly suitable for end point multiplex PCR. MyTaq HS Mix contains all the reagents (including stabilizers) necessary for trouble -free PCR set up. The product is supplied conveniently all in one tube to reduce the number of pipetting steps and to facilitate increased efficiency, throughput and reproducibility.ComponentsWebsite:/email:****************************PI -50152 V7Colony PCR ProtocolMyTaq HS Mix can be used for amplification of plasmid DNA directly from liquid cultures or from colonies on agar plates:- From liquid culture: up to 8 μL of the overnight culture can be directly added to the final reaction mix.- From colonies: we recommend using a sterile tip to stab the colony and resuspend it directly in the 50 μL reaction mix.Recommended cycling conditions for colony PCRof fragment up to 1 kb * These parameters may require optimization, please refer to the ImportantConsiderations and PCR Optimization section if needed.Multiplex PCR ProtocolMyTaq HS Mix is suitable for multiplex PCR; adjustment of the cycling conditions may be required. As a starting point we recommend using the following conditions:Recommended standard cycling conditions for multiplex PCR* These parameters may require optimization, please refer to the ImportantConsiderations and PCR Optimization section if needed.Important Considerations and PCR OptimizationThe optimal conditions may vary from reaction to reaction and are dependent on the template/primers used.Primers: Forward and reverse primers are generally used at the final concentration of 0.2-0.6 μM each. As a starting point, we recommend using a 0.4 μM final concentration (i.e. 20 pmol of each primer per 50 μL reaction volume). Too high a primer concentration can reduce the specificity of priming, resulting in non -specific products.When designing primers we recommend using primer -design software such as Primer3 (/primer3) or visual OMP TM () with monovalent and divalent cation concentrations of 10 mM and 3 mM respectively. Primers should have a melting temperature (Tm) of approximately 60 °C.Storage and stability:MyTaq HS Mix is shipped on dry/blue ice. On arrival store at -20 °C for optimum stability. Repeated freeze/thaw cycles should be avoided.Expiry:When stored under the recommended conditions and handled correctly, full activity of the kit is retained until the expiry date on the outer box label.Safety precautions:Please refer to the material safety data sheet for further information.Quality control specifications:MyTaq HS Mix and its components are extensively tested for activity, processivity, efficiency, heat activation, sensitivity, absence of nuclease contamination and absence of nucleic acid contamination prior to release.Notes:For research or further manufacturing use only.Trademarks:HyperLadder and MyTaq are trademarks of Bioline Reagents Ltd.Website:/email:****************************Troubleshooting GuideTemplate : The amount of template in the reaction depends mainly on the type of DNA used. For templates with low structural complexity,such as plasmid DNA, we recommend using 50 pg -10 ng DNA per 50 μL reaction volume. For eukaryotic genomic DNA, we recommend a starting amount of 200 ng DNA per 50 μL reaction, this can be varied between 5 ng -500 ng. It is important to avoid using templateresuspended in EDTA -containing solutions (e.g. TE buffer) since EDTAchelates free Mg 2+.Initial denaturation: The initial denaturation step is required to activatethe enzyme and fully melt the template. We recommend 1 minute of initial denaturation at 95 °C, however for more complex templates suchas eukaryotic genomic DNA, longer initial denaturation times of up to 3 minutes may be required.Denaturation: Our protocol recommends a 15 s cycling denaturationstep at 95 °C, which is also suited to GC -rich templates (>55%). For lowGC content amplicons (40-45%), the denaturation step can be decreased to 5 s.Annealing temperature and time: The optimal annealing temperatureis dependent upon the primer sequences and is usually 2-5 °C belowthe lower Tm of the pair. We recommend starting with a 55 °C annealingtemperature and, if necessary, running a temperature gradient to determine the optimal annealing temperature. Depending on the reaction the annealing time can also be reduced to 5 s.Technical SupportIf the troubleshooting guide does not solve the difficulty you are experiencing, please contact your local distributor or our Technical Support with details of reaction set -up, cycling conditions and relevant information.Email: ********************************Extension temperature and time: The extension step should beperformed at 72 °C. The extension time depends on the length of the amplicon and the complexity of the template. An extension time of 10 s is sufficient for amplicons under 1 kb or up to 5 kb for low complexity template such as plasmid DNA. For amplification of fragments over 1kbfrom high complexity template, such as eukaryotic genomic DNA, longer extension times are recommended. In order to find the fastest optimal condition, we suggest increasing the extension time up to 30 s/kb.Multiplexing : When doing multiplex PCR the recommended 2-stepcycling protocol may be optimized as follows:- Annealing/extension temperature: we highly recommend initially usinga temperature gradient to determine the optimal annealing temperature needed for the primer set used.- Annealing/extension time: in most cases a 4 min annealing/extensionstep is largely sufficient. However in order to reduce the overallcycling time this step can be reduced down to 1 min, especially in the case of a lower number of multiplex amplicons.- Cycling number: we recommend starting with 25 cycles and if necessary, optimizing this parameter. An excess of cycles may generate diffuse bands, too few may result in weak or no amplification. Associated Products ____________________________________________________________________________________________________________________________Bioline Reagents Ltd UNITED KINGDOMTel: +44 (0)20 8830 5300 Fax: +44 (0)20 8452 2822Meridian Life Science Inc. USATel: +1 901 382 8716 Fax: +1 901 382 0027Bioline GmbH GERMANYTel: +49 (0)3371 60222 00 Fax: +49(0)3371 60222 01Bioline (Aust) Pty. Ltd AUSTRALIATel: +61 (0)2 9209 4180 Fax: +61 (0)2 9209 4763。

Highly Available Long Running Transactions and Activities for J2EEApplications∗Francisco P´e rez-Sorrosal1,Jaksa Vuckovic2,Marta Pati˜n o-Mart´ınez1,Ricardo Jim´e nez-Peris1 1Universidad Polit´e cnica de Madrid,Spain2Universt`a di Bologna,Italy{fpsorrosal,mpatino,rjimenez}@fi.upm.es vuckovic@cs.unibo.itAbstractToday’s business applications are typically built on topof middleware platforms such as J2EE and use transactionsthat have evolved into long running activities able to adaptto different circumstances.Specifications,such as the J2EEActivity Service,have arised for applications requiring thatsupport.These applications also demand high availabilityto preventfinancial losses and/or service level agreements(SLAs)violations due to service unavailability or crashes.Replication is a means to attain high availability but currentmiddleware does not provide highly available transactions.In the advent of crashes,running transactions abort andthe application is forced to re-execute them,what results ina loss of availability and transparency.Most approachesusing J2EE consider the replication of either the applica-tion server or the database.This results in poor availabilitywhen the non-replicated tier crashes.This paper presents anovel J2EE replication support for both,application serverand database layers providing highly available transactionsand long running activities.Failure masking is transparentto client applications.A prototype has been implementedand evaluated.1IntroductionThe increasing degree of sophistication and complex-ity of modern business applications is demanding supportforflexible long running activities.Due to the long run-ning nature of business activities,traditional ACID transac-tions are not adequate for them.Several specifications havebeen proposed in the last years such as the activity servicefor CORBA[29]and J2EE[33],WS-Coordination/WS-Transaction[22]and WS-CAF[25].They provide the in-contribution consists in combining the replication of the ap-plication server(AS)and database tiers.This makes our platform resilient to any single point of failure.What it is more,it also guarantees the consistency between the two tiers in any failure scenario.The combination of the repli-cation of both tiers is especially novel in that replication is not made along tiers(i.e.replicating the AS tier indepen-dently of the database),but across them(i.e.replicating pairs of AS and database).The former approach is far from being trivial and requires some sophisticated logic to enable the consistent integration of both replicated tiers[20].The latter approach,the one taken in this paper,enables an in-tegral replication solution fully achieved at the AS tier and without modifying the database.This fact is important for pragmatic reasons since it enables the use of the replication platform with any existing database and without requiring access to the database code.A second contribution lies in that our solution supports highly available transactions and long running activities.Replica failures are transparently masked in front of clients.That is,running transactions and activities are not aborted.Upon failover,processing con-tinues at the point the primary was when it failed.All pre-vious solutions(that did not require specific hardware such as Tandem systems),to the best of our knowledge,abort ongoing transactions failing to satisfy the high-availability requirement.The provision of high availability in this con-text raises many challenges.Unlike previous work,it has to deal with services used by the application(e.g.the transac-tion manager,the activity service engine,etc.),in addition to business components.We have implemented and evaluated a prototype inte-grated into the JBoss AS[18].We have also implemented a prototype of the J2EE Activity Service for long running activities.The performance has been evaluated both with the ECPerf benchmark and a custom benchmark for long running activities.The paper is structured as follows.Section2intro-duces J2EE.Section3defines the system model.Section 4presents our replication algorithms.They are evaluated in Section5.Section6presents related work.Finally,Section 7concludes the paper.2J2EEJ2EE[32]is an extensible framework that provides a dis-tributed component model along with other useful services such as persistence and transactions.J2EE components are called Enterprise Java Beans(EJBs).There are three types of EJBs:session beans(SBs),entity beans(EBs)and mes-sage driven beans(MDB).We will not consider MDBs in this paper.SBs represent the business logic and their life-time is bounded to the lifetime of clients(i.e.,they are volatile).SBs are further classified as stateless(SSBs)and stateful(SFSBs).SSBs do not keep any state across method invocations.SFSBs may keep state across invocations of a client(conversational state).EBs model business data and are stored in some persistent storage,usually a database. The transaction manager is the service that handles trans-actions in J2EE.J2EE provides the Java Transaction API (JTA)to demarcate transactions.The transactional at-tributes of an EJB indicate whether its methods must run in a transaction,if a transaction must be already running or if a new transaction must be initiated.Traditional transactions may be too restrictive for appli-cations that need to relax some of the ACID properties.For instance,workflows may last for long periods of time(days, months).In this context,keeping locks across invocations drastically reduces the system concurrency.For this reason, several advanced transaction models have been proposed [10].The J2EE Activity Service specification(J2EEAS)al-lows the implementation of advanced transaction models in J2EE.The J2EEAS consists of two components:the activity service itself and one or more high level services(HLSs) (Fig.1).The activity service provides an abstract unit of work called activity,that may or may not be transactional. An activity may encapsulate a transaction or be encapsu-lated by a transaction.Activities may be nested.HLSs are defined on top of the activity service and repre-sent advanced transaction models.Applications use a HLS to demarcate activities,which produce an outcome.In or-der to implement a transaction model using the J2EEAS de-velopers must provide the demarcation points(signals),the outcomes and the state transitions(signalset).Figure1.J2EE services and componentsWe have implemented the open nested transactions model(ONT)[34]as defined in[33].ONTs are similar to traditional nested transactions(as specified in J2EE).How-ever ONTs relax the isolation property of nested transac-tions in order to be used in the context of long running transactions.In this way,a workflow can be seen as an atomic unit of work that is divided into smaller activities which are ACID transactions.When a subtransaction com-mits,all the resources are released.Due to the relaxation ofisolation,if the global activity fails,atomicity is achieved by logically undoing (compensating)committed activities.In order to compensate a committed transaction,when that transaction commits,it registers a compensator with its par-ent.This process is done recursively.When the top-level ONT commits,the registered compensators are discarded.If the top-level ONT aborts,compensators are applied in reverse order of completion.Figure 1shows the possible interactions among a client,EJBs,the transaction manager (TM),the ONT HLS and the activity service.3ModelIn our model,a replica is the pair AS and database.That is,ASs do not share the database.The set of all replicas is called a cluster (Fig.2).We will consider that a replica fails if either the database or the ASfails.Figure 2.Replication modelASs communicate using a group communication system (GCS)supporting strong virtual synchrony [14].GCSs pro-vide reliable multicast and group membership [8].Group membership services provide the notion of view (currently connected and active group members).Changes in the com-position of a view (member crash or new members )are eventually delivered to the application.We assume a pri-mary component membership [9].In a primary component membership,views installed by all members are totally or-dered (there are no concurrent views),and for every pair of consecutive views there is at least one member that sur-vives from the one view to the next one.Strong virtual syn-chrony ensures that messages are delivered in the same view they were sent (sending view delivery)and that two mem-bers transiting to a new view have delivered the same set of messages in the previous view (virtual synchrony).Group communication primitives can be classified attending to the order guarantees and fault-tolerance provided.FIFO order-ing delivers all messages sent by a group member in FIFO order.With regard to reliability,reliable multicast ensures that all available members deliver the same messages.Uni-form reliable multicast ensures that a message that is deliv-ered by a member (even if it fails),will be delivered at all available members.We assume that multicast messages are delivered to the sender of the message (self delivery).In the algorithms presented in this paper,ASs communicate using uniform FIFO multicast.4J2EE ReplicationIn this section we present a suite of replication algo-rithms for high available transactions (HA replication).The proposed algorithms follow a primary-backup replication scheme and take care of both the state of the AS and the database.That is,there is one replica (primary)that pro-cesses client requests and sends the state changes (check-point)to the rest of the replicas (backups).Backups just apply the changes the primary sends.Clients invoke EJBs residing on the primary and EJBs may invoke other EJBs.If the primary fails (either the AS or the database),a backup will take over and become the new primary.Clients will now interact with the new primary.Although we target long running transactions,first we present a replication algorithm for transactions whose life-time is a single client invocation to simplify the presen-tation.Then we will extend this algorithm with client-demarcated transactions that may invoke several times the AS within a single transaction,and finally,we will see how to replicate long running activities based on ONTs.The HA replication algorithms provide the following con-sistency properties in the absence of catastrophic failures (all replicas fail):1)Exactly once execution .Every re-quest is processed exactly once despite replica failures.2)Replica state consistency .After a client receives a reply,it is guaranteed that all running replicas have the same state.That is,the backups contain the same set of checkpoints and if the failover is performed,the backups will reach to the same state that the primary had (the same SFSBs with the same state,the same committed EB updates,the same database committed state,the same uncompleted transac-tions and uncompleted activities with the same associated updates and reads,and the same activity trees).3)Highly available processing .Every client request eventually re-ceives its outcome despite replica failures.That is,from the client perspective,transactions and activities never abort due to replica failures.It should be highlighted that the first and second proper-ties are provided by previous work,but only in the context of a single replicated tier (the application server tier).Our proposed algorithms fulfill these two properties replicating both tiers and therefore,without exhibiting any single point of failure.More importantly,our approach is the only one that fulfils the third condition to the best of our knowledge.4.1One Request TransactionsIn this algorithm we only consider transactions that start and complete during a client invocation to an EJB.This EJB may invoke other EJBs.The transaction will commit or abort before the client invocationfinishes.The client as part of the algorithm sends in each invo-cation a request identifier(rid).Rid consists of the client identifier and a request number.There is a request number per client that is increased each time that client invokes an EJB.This is done transparently by the client stub,so the client application code is not changed.When the primary receives an EJB invocation from a client,it starts a transaction and executes that request.This request may call other EJBs,resulting in a nested invoca-tion.The primary collects all the changes done to the in-voked EJBs(checkpoint)and the associated rid in a table (changes table)whose key is the client id.Request changes also include the creation and deletion of EJBs.Before re-turning the results to the client,the primary commits the transaction and FIFO multicasts to the backups the changes and the results of the request.Therefore,at most one mes-sage is sent to the backups per client invocation.If no EJB is modified,the primary does not send any message.The pri-mary returns the result to the client after it delivers the mul-ticast message with the EJB changes,if any.Uniform mul-ticast guarantees that if the primary has delivered that mes-sage,the backups will also deliver it.The primary commits transactions sequentially and FIFO multicasts the changes in the commit order to the backups.This guarantees that backups will commit transactions in the same order as the primary.If the transaction commits,the backups will re-ceive the committed EJBs changes and apply them.If the transaction aborts,no message corresponding to that trans-action is sent to the backups.Backups process primary mes-sages(applying the changes)in the delivery order(FIFO). Backups store the request identifier and the results in a ta-ble(results table).A backup keeps the results of a request message till it receives a new request from the same client. Failures.If an AS fails,the GCS informs all the ASs about the failure delivering a new view.If a backup has failed(it does not appear in the new view),nothing is done.If the primary fails,one of the backups will be chosen determin-istically to become the new primary(e.g.the replica with the smallest identifier in the view).Since strong virtual syn-chrony is assumed and messages are uniform multicast,all the backups that belong to the new view have delivered the same set of messages the old primary delivered when the new view is delivered.So,all of them will have the same EJB changes and anyone can take over as the new primary. If a database fails,the AS connected to it will detect the failure and shut down itself.Therefore,it will also fail.The new primary must apply the changes received from the old primary before starting to process client requests in order to ensure it has the same state the old primary had when it failed.If those changes are not applied by the time the view change is delivered,the processing of new client requests will be delayed until all outstanding changes are applied. In order to be able to fail over,the client stub needs to know the available ASs.The client stub receives the view of the cluster from the primary.Each view has a unique identifier(view id).The client stub attaches the view id to each request.If the primary detects that the client view id is not the id of the current view,it will piggyback the new view to the reply.So,if the primary fails,a client invocation will not succeed and the client stub will try to contact another AS from the last view received.That replica may or may not be the new primary.If the chosen AS is the new primary,it will process the request;otherwise it will reply with a message indicating the new primary.If the primary fails while processing a request,the client stub will receive an exception and resubmit that request to another server.Due to the use of uniform multicast,if the primary sent the changes before failing and delivered that message,the rest of the replicas also delivered the message and have the request changes and results.If the primary did not deliver that message,uniformity guarantees that the rest of the replicas will either all deliver the message or none.In the former case,the new primary has delivered the changes produced by that request.When the client resubmits the re-quest,the new primary will access the results table using the rid,detect that the request is a duplicate(the rid is in the results table)and send the cached reply to the client.In the latter case,none of the backups is aware of that request be-cause the primary failed before the corresponding changes were delivered.So,the new primary will access the results table and notfind that rid,and therefore will execute the re-quest.Otherwise,all of them are aware of it and then the behaviour is as in the former case.Note that the primary does not send any message to the backups if a request does not update any SFSB or EB (“read-only”request since SSB changes are not stored). Therefore,when a client resubmits a“read-only”request, the new primary is not aware of that request(the rid is not stored in the results table).The new primary will execute the request and return the results to the client.4.2Client-Demarcated TransactionsClients may demarcate transactions in J2EE using the Java Transaction API(JTA).A client-demarcated transac-tion may bracket several invocations to EJBs.Transactions are started,committed or aborted by the server transaction manager on behalf of the client.When a client begins a transaction,the primary will create a transaction and gen-erate a transaction identifier(tid).The primary stores thistid and the corresponding client identifier in a transaction table to associate that client with that transaction.Then, it multicasts this information(begin message)to the back-ups and suspends the transaction before returning to the client.The client attaches the tid and a request number to each EJB invocation from that transaction.When the pri-mary receives a request within that transaction,it accesses the transaction table to resume the associated transaction and then processes the request.The primary processes re-quests as in the basic algorithm(the primary stores the EJB changes,multicasts them together with the request results) but,before returning to the client it suspends the associated transaction.This transaction suspension just deassociates the thread from the transactional context(this is in fact done by applications servers even if they are not replicated).It has to be noted that now the primary sends committed and uncommitted changes to the backups.The state sent consists of the SFSBs and EBs modified in the current in-vocation.The state of SFSBs is not transactional.Even if a SFSB method runs within a transaction,if the transac-tion aborts that state is not undone to the previous state. Therefore,we will consider those changes as committed changes.When the client calls the commit(abort)oper-ation,the primary will resume the associated transaction, invoke the commit(abort)operation,and multicast a com-mit(abort)message with the tid to the backups.If SFSBs implement the afterCompletion method,the container will execute that method after committing(aborting)the trans-action.That method may change the state of the EJB.In this case,the primary will also send those changes in the commit(abort)message.An EJB method may start a new independent transaction when it is invoked.In this case,the enclosing(client)trans-action is suspended,and a new inner transaction is started. That is,in J2EE it is possible to run several transactions on behalf of a single client invocation,by suspending/resum-ing transactions.This implies that a message to a backup can carry changes from several transactions.When each client request corresponds exactly to a single transaction,backups only need to apply committed changes. They do not need to know which reads were performed in the database.However,when transactions span multiple client requests,this is not the case anymore.Reads per-formed by uncommitted transactions at the primary are im-portant to guarantee consistency if the primary fails,since they affect the serialization of transactions(i.e.by setting read locks).For instance,if there are two transactions T1 and T2,T1has read object a and T2has begun.Then,the primary fails.T1and T2are recreated on the new primary. Now,T2modifies object a and commits.T1performs other operations and reads a again.The value of a is different to the value read previously,though,both reads are executed within the same transaction(non-repeatable reads).If the primary would not have failed,T2would block when try-ing to modify a till T1finishes.In order to implement the same semantics for failure and failure free scenarios,the primary in addition to the changes also sends information about database reads.Since results of reads can be very bulky,the primary sends the SQL read statement submitted by the container to the database.When a backup receives a begin transaction message,it just stores the information received in the transaction table. Upon a backup receives changes from the primary,it ap-plies all committed changes and stores the request results in the results table.A backup will delay the application of uncommitted changes till it processes the commit message. When a backup processes the commit message,it applies the uncommitted changes for that transaction,discards the reads associated to the transaction,and stores the result of the transaction(commit)in the result table.If a backup re-ceives an abort message,it discards uncommitted changes and reads.Failures.If the primary fails before a client transaction completes,the new primary will recreate that transaction. The new primary will not process any client request till it has applied all the messages sent by the old primary.When this happens,the primary checks if there are uncompleted transactions in the transaction table.If this is the case,the new primary will recreate a transaction for each of these transactions,apply uncommitted changes and execute the associated reads in the order they were sent by the old pri-mary.This guarantees that each recreated transaction will hold the same state it held at the old primary,therefore guar-anteeing consistency of recreated transactions.If the primary fails between two client invocations,the client just needs to contact the new primary.If the primary fails while running an invocation,the client stub will resub-mit the invocation behaving as in the previous algorithm. If the primary fails when the client called the begin opera-tion,there are two cases.If the transaction is in the trans-action table,the new primary will send back the tid.Oth-erwise,the new primary did not receive the begin message in the previous view and will process it now.Finally,if the commit(abort)operation failed,the new primary might have the result and return it to the client,or not have it,in which case it will execute the operation.Replicating the un-committed state on each client invocation avoids the abor-tion of the client transaction in case the primary fails.The new primary resumes the transaction transparently provid-ing highly available transaction support.4.3Open Nested TransactionsLike client-demarcated transactions,ONT activities are started,completed and compensated on the server and may involve multiple server invocations.ONT activities are de-marcated either by the client or by SBs.This can yield to arbitrarily complex ONT activity trees as it happens with nested transactions in JTA.The main difference from the replication point of view is that whenever a child ONT ac-tivity succeeds,a compensator is registered with its parent. This procedure is performed recursively until the top-level ONT activity is reached.When the top activity commits, registered compensators are discarded.Whenever an ONT activity aborts,the registered compensators are executed. Therefore,in order to replicate ONT activities and provide transparent failover the primary must send the correspond-ing ONT activity tree state with all this information to the backups.When a client starts an ONT activity,the primary gen-erates an activity identifier(aid)and stores it with a client identifier in an activity table.The primary multicasts an ac-tivity begin message to the backups with that information, suspends the ONT activity and returns the aid to the client. When a client submits a request,it attaches the aid and a request identifier.The primary resumes the associated ONT activity and executes the request.That request may start a nested ONT activity.The primary will register that ONT activity in the ONT activity table as a child ONT activity of the client ONT activity(ONT activity tree).If the nested ONT activity succeeds,a compensator is registered with the parent ONT activity to be invoked in case the parent ONT activity fails.The primary will multicast the request identi-fier,the request result,the changes of the completed nested ONT activity as committed changes,the uncompleted par-ent ONT activity changes as uncommitted ones,read state-ments,the ONT activity tree with the status of each ONT activity in the tree and the registered compensators before returning to the client.In the case under consideration,the child activity has committed and therefore,the ONT activ-ity tree has a single ONT activity(the parent).When the client submits a commit operation,the primary resumes and commits the associated ONT activity,multicasts the ONT activity outcome,and returns to the client.Now the aid is removed from the ONT activity table.If the parent ONT activity fails,the primary executes the compensators and af-terwards,it collects all the EJB changes.Then,it multicasts the changes and the ONT activity outcome.After delivering this message,it returns the outcome to the client. Backups store the aid and the client id when they re-ceive a begin message.When a backup receives a message with changes,it updates the ONT activity tree according to the one in the message and proceeds as in the previous algorithm.Uncommitted changes will be applied when a commit message is received,and discarded if the message is abort.In that case,the compensators are executed. Failures.When the new primaryfinishes processing mes-sages from the previous primary,it reconstructs all unfin-ished ONT activities.For each ONT activity in the table,it traverses top-down the corresponding ONT activity tree. The new primary creates an(possibly nested)ONT activ-ity and associates the registered compensators,if any,for each node in the ONT tree.Then,it applies uncommitted changes,and executes the reads performed on behalf of the ONT.Now,the primary can resume request processing.5EvaluationWe have implemented the replication algorithms in the JBoss AS.Our implementation is based on the ADAPT framework[2].It supports the prototyping of replication protocols in JBoss using interceptors.The ADAPT frame-work provides two types of interceptors:client component monitor(CCM)and component monitor(CM).The client component monitor intercepts all the outgoing invocations at the client side.It implements the client side of the repli-cation algorithms.The CCM is dynamically loaded by the client when getting the stub of a remote EJB.There is one CCM instance per client.The CM is the server side coun-terpart responsible for intercepting both,remote invocations from the client to the EJBs,and local invocations between EJBs.It implements the server side of the replication algo-rithm.We have evaluated the overhead of the replication algo-rithms both in a traditional transactional application and in an application based on the ONTs model.We used a non-replicated JBoss and JBoss clustering as baselines for the experiments.Although,none of them provides the avail-ability and fault-tolerance guarantees our algorithms do.In JBoss clustering there is a single shared database.The repli-cation algorithm of JBoss is not transaction aware and trans-fers the state of an SFSB at the end of each invocation that modifies the SFSB.JBoss clustering is configured to exe-cute as a primary-backup.We have also measured the time needed for failover when the primary fails.The experiments were run in a cluster of2GHz AMD dual-processor PCs with512MB of RAM running Red Hat Linux9.0.We used JBoss3.2.3AS[18],PostgreSQL7.3.2 database,JGroups[1]as a GCS and JASS[26],our imple-mentation the activity service.In all experiments the clients, each instance of JBoss and the database were run in separate hosts.All the results of the experiments have been obtained over the steady phase of the test.5.1Transaction ReplicationThe evaluation of the replication algorithm has been done using ECperf[31].It simulates a supply chain,defin-ing four application domains:corporative,order entry,sup-ply chain management and manufacturing.ECperf mea-sures the throughput in BBops per minute(benchmark busi-ness operations).BBops are the sum of the number of trans-。