侯柱峰-vasp计算晶体弹性常数
- 格式:pdf
- 大小:346.90 KB
- 文档页数:7


VASP表面计算步骤小结(侯博士)一、概述vasp用“slab”模型来模拟表面体系结构。
vasp计算表面的大概步骤是:材料体性质的计算;表面模型的构造;表面结构的优化;表面性质的计算。
二、分步介绍1、材料体性质计算:本步是为了确定表面计算时所需的一些重要参数:ENCUT、SIGMA(smearing 方法为ISMEAR=1 或0时;而通常表面体系结构优化时选择这种smearing方法)、晶格参数。
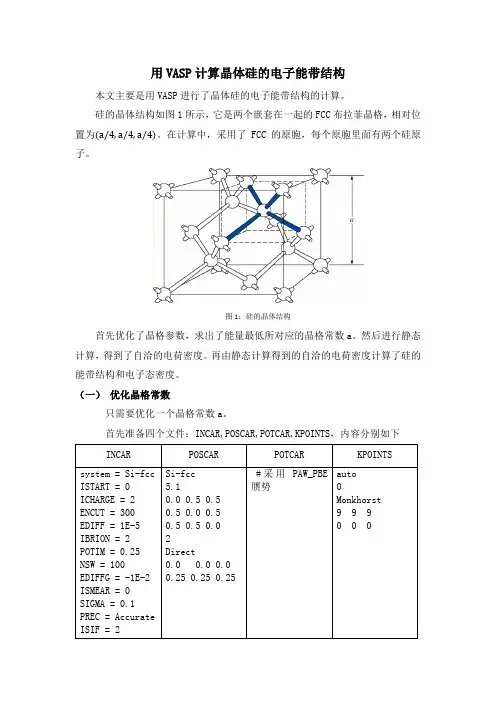
<一>在计算前,要明确:何种PP;ENCUT;KPOINTS ;SIGMA;PREC;EX-CO,这其实是准备proper input files。
a. 何种PP选择的PP能使计算得到的单个原子能量值在1meV~10meV之间。
[参见P 21]所求得的单原子能量(对称性破缺时)可用来提高结合能的精度。
b. ENCUT [ 参见P 14 ]选择的ENCUT应使得总能变化在0.001eV左右为宜。
注意:试探值最小为POTCAR中的ENMAX(多个时,取最大的),递增间隔50;另外,在进行变体积的结构优化时,最好保证ENCUT=1.3ENMAX,以得到合理精度。
c. PREC [参见P 16]控制计算精度的最重要参数,决定了(未指定时)ENCUT、FFT网格、ROPT取值。
一般计算取NORMAL;当要提高Stress tensor计算精度时,HIGH 或ACCURATE,并手动设置ENCUT。
d. EDIFF & EDIFFG [参见P16]EDIFF 判断电子结构部分自恰迭代时自恰与否,一般取默认值=1E-4;EDIFFG 控制离子部分驰豫e. ISTART & ICHARGE [参见P 16]ISTART = 1, ICHARG = 11:能带结构、电子态密度计算时;ISTART =0, ICHARG = 2:其余计算ISTART = 1,ICHARG = 1(其他所有不改变):断点后续算设置f. GGA & VOSKOWN [参见P 16]GGA=91: Perdew -Wang 91;GGA=PE: Perdew-Burke-ErnzerhofVOSKOWN=1( GGA=91时);VOSKOWN=默认(其余情况)g. ISIF [参见P 16]控制结构参数之优化。

Next: 采用VASP如何计算晶体的弹性常数 Up: VASP计算实例 Previous: 用VASP 计算Pd金属的晶格常数 Contents用VASP 计算表面能做表面计算时,第一步我们需要测试K 点的收敛性。
通常,在垂直表面方向用1个K 点就可以了,在平行表面方向,可以用和体材料类似的K 点密度。
其次,我们要测试真空厚度(vacuum thickness)的收敛性。
我们构造完一个slab 后,将真空厚度逐渐从增加到,体系的总能量改变不超过10meV 的时候,可以初步认为真空厚度达到标准。
以下是一个3层的(fcc) Pd slab 的能量随着真空厚度的变化。
其INCAR 文件如下:SYSTEM = undeformed fcc Pd (111) surface calculation Startparameter for this run: PREC = AccurateISTART = 0 job : 0-new 1-cont 2-samecut ICHARG = 2 charge: 1-file 2-atom 10-const ISPIN = 1 spin polarized calculation?Electronic Relaxation 1NELM = 90; NELMIN= 8; # of ELM steps EDIFF = 0.1E-03 stopping-criterion for ELM LREAL = .FALSE. real-space projection NBANDS = 40Ionic relaxationEDIFFG = 0.1E-2 stopping-criterion for IOM NSW = 0 number of steps for IOMIBRION = 2 ionic relax: 0-MD 1-quasi-New 2-CG ISIF = 2 stress and relaxationPOTIM = 0.10 time-step for ionic-motion TEIN = 0.0 initial temperatureTEBEG = 0.0; TEEND = 0.0 temperature during runDOS related values:ISMEAR = 1 ; SIGMA = 0.20 broadening in eV -4-tet -1-fermi 0-gausElectronic relaxation 2 (details)Write flagsLWAVE = F write WAVECARLCHARG = F write CHGCARLVTOT = .TRUE.其中因为Pd是金属,ISMEAR设置为method of Methfessel-Paxton。


VASP计算弹性常数VASP (Vienna Ab-initio Simulation Package) 是一种常用的第一性原理计算软件包,用于计算物质电子结构和能带结构。
除了电子结构计算,VASP还可以用于计算材料的弹性常数。
在本文中,我们将讨论如何使用VASP计算材料的弹性常数,并了解计算结果的解释。
弹性常数是描述材料变形行为的物理量。
材料在受力作用下产生变形,而弹性常数则定量描述了材料对应力的响应。
弹性常数包括弹性模量、剪切模量、泊松比等。
通过计算这些弹性常数,我们可以了解材料的机械性能和应力应变行为。
首先,我们需要确定计算材料的晶体结构。
这包括晶胞参数、原子位置和晶胞对称性。
在VASP中,我们使用POSCAR文件来描述晶体结构的具体细节。
POSCAR文件包括晶体的晶胞参数、原子种类和位置等信息。
其次,我们需要生成一系列的应变状态。
常见的应变状态包括体积应变、晶格常数应变和剪切应变。
在VASP中,我们可以使用ISIF标志来控制应变类型。
例如,ISIF=3可以用于计算体积应变,ISIF=2可以用于计算剪切应变。
然后,我们需要进行一系列的弛豫计算。
在每个应变状态下,我们需要优化结构以达到最低的总能量。
这可以通过设置IBRION=2和ISIF=3来实现。
这些计算将给出最优的应变状态下的应力张量。
最后,我们可以使用应力和应变的关系来计算材料的弹性常数。
对于单晶材料,弹性常数可以通过应力张量的分量和应变张量的分量之间的线性关系来得到。
C_ij = (stress_i - stress_0_i) / strain_j其中,C_ij是第i个应力分量(i = 1,2,...,6)和第j个应变分量(j = 1,2,...,6)之间的弹性常数,stress_i是在第i个应变分量下计算得到的应力,stress_0_i是在未应变状态下计算得到的应力,strain_j是第j个应变分量。
使用以上计算方法,我们可以得到材料的弹性常数。

侯柱峰-vasp计算晶体弹性常数引言晶体弹性常数是描述晶体材料力学性质的重要参数之一,对于理解材料的力学行为和设计新材料具有重要意义。
在材料科学领域中,计算晶体弹性常数的方法多种多样,其中基于第一性原理的计算方法得到了广泛的应用和认可。
本文将介绍侯柱峰等人在VASP软件包中计算晶体弹性常数的方法和步骤。
VASP简介VASP(Vienna Ab-initio Simulation Package)是一款基于密度泛函理论(DFT)的第一性原理计算软件包,由维也纳大学的J. Hafner教授及其团队开发。
VASP以固体物理和材料科学为核心,广泛应用于研究材料的电子结构、力学性质、热力学性质等方面。
VASP计算晶体弹性常数的方法1. 结构优化在计算晶体弹性常数之前,首先需要通过VASP进行结构优化。
结构优化的目的是找到晶体的平衡结构,使得能量最低。
在结构优化过程中,需要设定材料的晶格参数、原子种类和初始位置等参数,并设置计算的精度和收敛标准。
2. 弹性常数计算前的准备在进行弹性常数计算之前,需要对结构进行静力学计算,即计算晶体的内部应力状态。
为此,需要在INCAR文件中设置一些相关的参数,例如选用的泛函类型、截断能等。
3. 弹性常数计算在进行弹性常数计算之前,需要在INCAR文件中添加以下参数:ISTART = 1ICHARG = 2ENCUT = 520ISYM = 0ISMEAR = 0然后运行VASP进行弹性常数的计算。
计算完成后,可以得到弹性常数矩阵,其中包括36个元素,分别对应不同的弹性常数。
4. 弹性常数的后处理在得到弹性常数矩阵之后,需要进行一些后处理来得到弹性常数的具体数值。
这一步可以使用第三方软件进行,例如使用MATLAB来计算并提取需要的弹性常数。
结论侯柱峰等人在VASP软件包中提供了一种计算晶体弹性常数的方法,可以方便而准确地得到晶体的力学性质。
这种方法基于第一性原理,充分考虑了材料的电子结构和原子之间的相互作用,具有很高的可靠性和适用性。

如何用VASP计算晶格常数VASP是一款常用的第一性原理计算软件,可用于计算各种物理和化学性质,包括晶格常数。
本文将通过详细的步骤指导如何使用VASP计算晶格常数。
1.准备工作:在使用VASP计算晶格常数之前,需要准备以下文件:-INCAR文件:包含所有计算参数的输入文件。
- POSCAR文件:包含体系的原子坐标和晶格常数的输入文件。
可以使用外部软件生成,例如Materials Studio、VESTA等。
-POTCAR文件:包含原子势能信息的文件。
-KPOINTS文件:用于定义k点网格,用于计算能带结构。
可以使用自动生成工具进行生成。
2.设置INCAR文件:打开INCAR文件,设置以下参数:-ENCUT:截断能。
一种势能截断参数,对计算结果影响较大。
可通过多次计算逐渐增大其值,直到结果收敛为止。
- ISMEAR:用于定义电子占据数的方法。
常用的选项有Gaussian和Methfessel-Paxton。
- SIGMA:在使用ISMEAR选项为Gaussian时,用于定义宽度的参数。
一般选择小于0.2 eV。
- PREC:定义计算的精度级别。
常用的设置有Low、Normal和High。
-NSW:定义离子进行多少步的迭代。
-ISTART和ICHARG:对于初始的计算,将其设置为0。
-EDIFF:收敛判据。
设置一个合适的值,使得计算结果收敛。
3.设置POSCAR文件:打开POSCAR文件,设置晶体的结构参数。
可以手动输入原子的坐标,或者复制其他软件生成的文件内容。
4.设置POTCAR文件:在VASP的安装目录中,找到POTCAR文件夹,并将需要使用的原子势能文件复制到当前工作目录中。
注意保持POTCAR文件的顺序和POSCAR文件中原子的顺序一致。
5.设置KPOINTS文件:打开KPOINTS文件,在其中设置k点的信息。
k点的密度对计算结果的精度有一定影响,可以根据具体需求进行调整。
在这里,我们将只计算晶格常数,因此可以选择较低的k点密度。

vasp计算弹性常数
VASP计算弹性常数
1、什么是VASP计算弹性常数
VASP是Vienna Ab initio Simulation Package(维也纳基础仿真软件包)的缩写,它是一种针对第一性原理计算的电子结构和性质的软件套件。
它在计算与物理、化学能量和结构有关的量时具有非常强大的能力,可以计算出材料的结构构型、热力学性质以及电学和光学性质等。
VASP也可以用来计算某个材料的力学性质,从而推断其弹性常数。
2、VASP计算弹性常数的原理
了解VASP计算弹性常数的原理,我们先需要了解Hooke定律。
Hooke定律定义了弹性体(指正交于拉伸方向的任意方向上发生同样变形的材料)应力和应变之间的关系,即:拉伸轴上拉伸应力可以由材料弹性常数通过应变除法表示。
为了计算材料的弹性常数,利用VASP软件可以通过模拟调节其立体结构的应力和应变,从而推断出材料的弹性常数。
它首先会对所模拟的晶体进行平衡计算,根据平衡晶体的初始位置和能量,将其一点点推向应变,然后根据新推向位置和能量,重新计算势函数,最后弹性常数就可以根据VASP软件计算出来。
3、VASP计算弹性常数的常用方法
4、VASP计算弹性常数的应用
VASP计算弹性常数有很多强大而精准的应用,相当多的材料比如金属、合金、多晶体以及纤维、木材和塑料等都可以通过VASP软件来计算它们的弹性常数,从而快速准确地预测出它们在拉伸、压缩、弯曲等应力下的反应情况。
此外,有了VASP的计算结果,我们也可以进一步提取出动态弹性常数,从而更好地了解材料的形变行为及表面粗糙度、能金学性质以及其他有关的研究。


vasp计算弹性常数
弹性常数(elasticmodulus)是材料力学中最重要的物理量之一,它表征材料的刚性程度。
它是描述材料弹性性质的唯一参数,也是物理化学中最常用的概念之一。
现在,它已成为材料设计和模拟的研究实验。
VASP(Vienna Ab initio Simulation Package)是一种非凡的
计算机软件,它可用于从原子层面来预测材料的性能。
其优点在于它可以有效计算和模拟态密度和势能,电子结构,以及其他物理性质,如弹性常数。
VASP计算弹性常数具有一定的复杂性。
它首先要求设定计算参数,以确定计算精度和计算极限。
其次,运行VASP软件,确定给定
温度和压力下材料的刚度,然后再根据极限状态或近似状态求取弹性常数。
最后,运行VASP以验证计算结果。
计算弹性常数的最终目的是确定材料的机械性质,以预测材料的整体性能。
这一过程可以利用VASP软件实现,它将原子中的电子结
构转化为材料的结构性质及弹性常数。
此外,VASP的计算速度也有
极大的优势,可以在可接受的时间内完成大量的计算任务,给常规材料设计和模拟计算提供重要的计算支持。
综上所述,VASP可以用来有效计算材料的弹性常数,并且这一
过程有着极大的优势,它可以在可接受的时间内完成大量的计算任务,给常规材料设计和模拟计算提供重要的计算支持。
因此,VASP可以
成为有效的材料设计和模拟工具,可以有效的计算和模拟材料的弹性
常数。
按照HfN的NaCl结构,弛豫结果:K点最优是21*21*21,ENCUT取550eV,sigma取0.2,EV优化的结果也很理想,最优体积为23.28854892,换算之后,晶格常数是4.533457483,与参考文献一致。
对原来的晶格基矢矩阵做形变[e=(r,r,r,0,0,0)],把形变后的矩阵(矩阵换算规则:R’=R*(δ+e))代替原矩阵放到POSCAR中:000#!/bin/sh#PBS -N vasp#PBS -j oe#PBS -l nodes=1:ppn=12cd ${PBS_O_WORKDIR}source /public/software/profile.d/intel-env.shsource /public/software/profile.d/openmpi-intel-env.shechoecho "Starting V ASP run at" `date`echomaster=`hostname`echo "The job submission node is $master"echo "The working directory is " ${PBS_O_WORKDIR}echo "V ASP input file is" ${PBS_O_WORKDIR}/${inputfile}echoecho "V ASP execution start at" `date`echohostname > grep 'Linux' /etc/issue >> grep 'model name' /proc/cpuinfo |cut -d: -f2 |uniq -c >> grep 'cpu M' /proc/cpuinfo >> grep 'MemTotal' /proc/meminfo >> free -g >> ulimit -a >> cat $PBS_NODEFILE >> NP=`cat $PBS_NODEFILE | wc -l`#################################################################rm W A VECAR 2>/dev/nullechofor i in -0.02 -0.018 -0.016 -0.014 -0.012 -0.01 -0.008 -0.006 -0.004 -0.002 0 0.002 0.004 0.006 0.008 0.01 0.012 0.014 0.016 0.018 0.02; dok=`echo $i | awk '{printf"%.7f",$i*0.5+0.5}'`cat > POSCAR <<!Cubic HfN4.533357680.000000000000000 $k $k 原始矩阵:0 0.5 0.5$k 0.000000000000000 $k 0.5 0 0.5$k $k 0.000000000000 0.5 0.5 01 1Direct0.00000000 0.00000000 0.000000000.50000000 0.50000000 0.50000000!cd /home/yjhao/chenlongqing/r/r1mpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=$i $E" >> TOTALcp CONTCAR POSCAR.$icp OUTCAR OUTCAr.$idonecp INCAR.static INCARcp POSCAR.-0.02 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.02 $E" >> TOTANcp POSCAR.-0.018 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.018 $E" >> TOTANcp POSCAR.-0.016 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.016 $E" >> TOTANcp POSCAR.-0.014 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.014 $E" >> TOTANcp POSCAR.-0.012 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.012 $E" >> TOTANcp POSCAR.-0.01 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outcp POSCAR.-0.008 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.008 $E" >> TOTANcp POSCAR.-0.006 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.006 $E" >> TOTANcp POSCAR.-0.004 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.004 $E" >> TOTANcp POSCAR.-0.002 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=-0.002 $E" >> TOTANcp POSCAR.0 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0 $E" >> TOTANcp POSCAR.0.002 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.002 $E" >> TOTANcp POSCAR.0.004 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.004 $E" >> TOTANcp POSCAR.0.006 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.006 $E" >> TOTANcp POSCAR.0.008 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.008 $E" >> TOTANcp POSCAR.0.01 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.01 $E" >> TOTANcp POSCAR.0.012 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outcp POSCAR.0.014 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.014 $E" >> TOTANcp POSCAR.0.016 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.016 $E" >> TOTANcp POSCAR.0.018 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.018 $E" >> TOTANcp POSCAR.0.02 POSCARmpirun -np $NP-machinefile $PBS_NODEFILE --mca btl self,sm,tcp -bind-to-core numactl --localalloc /public/software/vasp5.2/vasp >outE=`grep F= OSZICAR` ; echo "r=0.02 $E" >> TOTAN############################################################################### ##########3exit 0相对应的,TOTAN文件的内容如下:r=-0.02 1 F= -.21835920E+02 E0= -.21835920E+02 d E =0.000000E+00r=-0.018 1 F= -.21837975E+02 E0= -.21837975E+02 d E =0.000000E+00r=-0.016 1 F= -.21839808E+02 E0= -.21839808E+02 d E =0.000000E+00r=-0.014 1 F= -.21841417E+02 E0= -.21841417E+02 d E =0.000000E+00r= -0.012 1 F= -.21842791E+02 E0= -.21842791E+02 d E =0.000000E+00r= -0.01 1 F= -.21843936E+02 E0= -.21843936E+02 d E =0.000000E+00r=-0.008 1 F= -.21844865E+02 E0= -.21844865E+02 d E =0.000000E+00r=-0.006 1 F= -.21845578E+02 E0= -.21845578E+02 d E =0.000000E+00r=-0.004 1 F= -.21846085E+02 E0= -.21846085E+02 d E =0.000000E+00r=-0.002 1 F= -.21846387E+02 E0= -.21846387E+02 d E =0.000000E+00r=0 1 F= -.21846497E+02 E0= -.21846497E+02 d E =0.000000E+00r=0.002 1 F= -.21846390E+02 E0= -.21846390E+02 d E =0.000000E+00r=0.004 1 F= -.21846095E+02 E0= -.21846095E+02 d E =0.000000E+00r=0.006 1 F= -.21845602E+02 E0= -.21845602E+02 d E =0.000000E+00r=0.008 1 F= -.21844905E+02 E0= -.21844905E+02 d E =0.000000E+00r=0.01 1 F= -.21843996E+02 E0= -.21843996E+02 d E =0.000000E+00r=0.012 1 F= -.21842878E+02 E0= -.21842878E+02 d E =0.000000E+00r=0.014 1 F= -.21841544E+02 E0= -.21841544E+02 d E =0.000000E+00r=0.016 1 F= -.21840003E+02 E0= -.21840003E+02 d E =0.000000E+00r=0.018 1 F= -.21838261E+02 E0= -.21838261E+02 d E =0.000000E+00r=0.02 1 F= -.21836314E+02 E0= -.21836314E+02 d E =0.000000E+00将TOTAN中的r和E0提取出来,再用Origin来做拟合(阶数选4,提高精度)可以看到,B2结果是25.228367816,带入下式:V0=23.29163453C44=2B2*160.2/3V计算结果是C44=115,与文献数据相符同理,施加形变e=(0,0,0,r,r,r)和e=(r,r,0,0,0),大体类似,改变的只是变换后的矩阵,分别是:0.00 $k $k$k 0.0 $k$k $k 0.00和0.00 $k 0.50$k 0.0 0.50$k $k 0.00分别将TOTAN文件里面的能量提出,并且转换成,代入Origin里面进行拟合,得到的B2分别是186.4783989和106.79144这样计算之后:C11+2C12=855(这个数据除以3应等于B0,根据EOS拟合,B0=285,完全符合), C11+C12=734最后得出:C11=613,C12=121,C44=115与参考文献一致:。
VASP计算有限温度下的晶格常数一般VASP算得的晶格常数是在0k下的,现在如果要算300k下的晶格常数,用VASP可否实现。
一种近似是认为二者一样。
反正是在常温常压下,距离熔点很远。
一般来说,VASP计算不能加上温度,除非是做分子动力学的计算,来模拟固定在一定温度下稳定性。
要计算高温下固体的能量和晶格常数、体积,可以计算出不同晶格常数(也就是体积)固体的声子振动频率,再得到自由能,利用F(V)的关系得到在某一个温度下的平衡体积。
(这种办法是一种准谐近似),PRB 上面有很多类似的文章。
有人会问:(1)既然涉及到温度都只能是MD了,为何还能计算高温下的声子振动频率?答:这是因为MD计算也能求解出声子的振动频率,这个在Rev.Mod.Phys.2001的一篇文章就有提到。
(2)得到声子的振动频率在VASP中是设定IBRION=5,但是只能计算Gama 点的振动频率,你的意思是计算Gama点的频率?答:VASP可以任何k点的声子振动频率,不局限于Gamma点。
手册上有提到的,要计算其他非Gamm点的频率需利用一个PHONON的程序。
(3)用VASP计算后的OSZICAR中就直接输出自由能呀!为何还通过计算声子频率再得到自由能呢?怎样得到的呢?答:VASP输出的自由能只是电子部分的,非原子的自由能。
看看VASP 在comput.mat.sci.上面的一篇文章就知道了vasp的理论基础。
G. Kresse and J. Furthmüller, "Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set", Comput. Mat. Sci. 6, 15-50 (1996).如果希望采用准谐近似来计算热学性质,可以参看这样的一篇文章(尽管是对金属单质体系的)PHYSICAL REVIEW B, VOLUME 65, 064302,2002 Ab initiocalculation of the thermal properties of Cu: Performance of the LDA and GGA。
我们用Pd金属作为例子。
Pd金属的实验上的晶格常数为3.89A。
在这里,我们用V ASP计算它的晶格常数。
首先将Pd所对应的POTCAR文件拷贝到目录下。
然后准备好INCAR和KPOINTS文件。
POSCAR文件我们将通过一个tcsh的script来产生。
KPOINTS文件可以如下:Monkhorst PackMonkhorst Pack11 11 110 0 0INCAR文件可以如下:SYSTEM = Pd bulk calculationStartparameter for this run:PREC = AccurateISTART = 0 job : 0-new 1-cont 2-samecutICHARG = 2 charge: 1-file 2-atom 10-constISPIN = 1 spin polarized calculation?Electronic Relaxation 1EDIFF = 0.1E-03 stopping-criterion for ELMLREAL = .FALSE. real-space projectionIonic relaxationEDIFFG = 0.1E-02 stopping-criterion for IOMNSW = 0 number of steps for IOMIBRION = 2 ionic relax: 0-MD 1-quasi-New 2-CGISIF = 2 stress and relaxationPOTIM = 0.10 time-step for ionic-motionTEIN = 0.0 initial temperatureTEBEG = 0.0; TEEND = 0.0 temperature during runDOS related values:ISMEAR = 0 ; SIGMA = 0.05 gaussian smearElectronic relaxation 2 (details)Write flagsLWA VE = F write WA VECARLCHARG = F write CHGCAR产生POSCAR和计算晶格常数的工作可以用以下的PBS script来完成。
式中C ij就是我们通常所说的弹性模量,可以证明,上述刚度矩阵为对称阵,C ij=C ji,因此,弹性模量的独立张量元数目至多只有21个。
晶系的对称性越高,独立的张量元数目就越少。
需要指出的是,C ij的数目只与晶系有关,而与晶系中具体的对称类型无关。
下面分别讨论七种不同晶系的弹性模量矩阵元:
1.1 三斜晶系(Triclinic system)
三斜晶系是所有七大晶系中对称性最低的晶系,因此拥有最多的独立矩阵元,其形式为:
从上式可以看出,正交晶系拥有相当高的对成性,其独立刚度矩阵元的数目为8
三角晶系(32,3m,32/m)的独立刚度矩阵元的数目为6个。
立方晶系是所有晶系中对称度最高的晶系,其独立的刚度矩阵元数目仅为3个。
至此,我们列出了所有七大晶系的刚度矩阵元,只要求出各晶系对应的所有独立矩阵元,即可得到晶体的刚度矩阵。
V ASP的个人经验手册使用V ASP的个人经验手册侯柱锋厦门大学物理系2004届博士E-mail: zhufhou@ 2004/06/22 本手册纯属个人使用V ASP后的心得和经验总结,版权属于本手册的作者及厦门大学物理系计算物理实验室。
未经许可,不准在网上传阅。
文中提到的一些小程序,可以提供使用。
在参考的过程,如遇到不清楚或含糊的地方,可以参考V ASP的英文manual或email给我。
如认为本手册某些地方需要更正或修改的,请email给我。
当在使用V ASP的过程中遇到问题,也可以email给我,大家一起学习V ASP 的使用,挖掘和掌握V ASP强大的功能。
本手册参考了V ASP的英文manual、的报告以及从internet网上收集的资料。
本手册大致有以下几个内容:A程序的编译“?A V ASP的主要输入文件OA V ASP的主要输出文件lA参数设置与选择的技巧A材料基态性质的计算方法和步骤ZA材料磁性性质的计算μA表面体系的计算”aAtools中小程序的说明A半导体中的缺陷和杂质问题十、如何进行分子动力学模拟十一、强关联体系的计算(LDA+U或GGA+U) 一、程序的编译声明:本实验室购买的是版本,所属本实验室的成员以及经过朱梓忠教授同意使用的合作者必须遵守该软件的使用协议,注意V ASP软件的版权问题,严禁私下发布或传播本实验室购买的V ASP源代码和赝势库以及编译V ASP得到的可执行代码。
1下面以编译版本为例,编译更新的版本、和的步骤与此相同。
1、所需文件和程序V ASP源代码:和数学库:LAPACK和BLAS (/),或mkl,或ATLAS (/) 或Lib GOTO (/users/flame/goto/) Fortran编译器:PGI fortran 至少以上版本(/),或Intel的ifc (以上版本是ifort, /software/products/compilers/flin/),前者可以从网站上下载到15天的试用版本,后者可以从网站下载到的版本。