A silica monolithic column prepared by the sol-gel process for enantiomeric separation by
- 格式:pdf
- 大小:112.02 KB
- 文档页数:5

The Company: BIA SeparationsThe Technology: CIM Convective Interaction Media®Rapid Method Development&Analyticswith Convective Interaction Media®10–32 VALCO-type connectors, the column can be fitted to any LC, HPLC, Disks are easily placedinto the housing allowing simple column handling; fast,two different plastic versions: POM (blueaccommodate up to 4By packing the same chemistry, the volume (capacity) andlength can be increased; by packing different chemistries,Simple Scale-upNote: The Chromatogram for the 8000 ml column is an extrapolation From: Milavec @mak et al., J. Chrom. A, 2003, 1006, 195Once you have developed your method on a CIM ®disk monolithiccolumn, you can easily scale it up to the preparative or industri-al scale. With a range of available volumes up to 8liters, you aresure to find the right column for your process. Furthermore,scale-up is straightforward since all columns perform like the CIM ®disk monolithic columns. As shown below, the resolution is pre-served regardless of the column used. The columns are designed to meet the most stringent demands of biochemists and process engineers; operating pressures from 10 to 50bar, temperatures up to 50°C, pH 1–14 and they can be used with all common polar solvents. All preparative and industrial scale CIM ®columns are supplied in ready to use housings (cGMP compliant if requested).Column figures are not to scale!CIM®tube monolithic column structureThe preparative and industrial scale CIM®tube monolithic columnsare typically placed on an inlet plate and have a frit on the inner side.Two Teflon seals placed on the top and bottom of the monolith ensuresealing, while the whole monolith is covered by the column body andoutlet plate. CIM®tube monolithic columns are designed for simplehandling and robust processing and guarantee customer satisfaction.1234567 inlet plate bottom Teflon®seal frit monolith column body top Teflon®seal outlet plate✸✸✸✸✸✸✸✸✸✸✸✸12345Plasmid DNA – characteristics and challengesPlasmid DNA is a closed-loop double stranded DNA that occurs naturally in bacteria. They are very suitable vectors (delivery vehicles) for gene therapy applications where highly pure pDNA is needed. It must be virtually free from impurities like genomic DNA, RNA,proteins and endotoxins and >90% must be in the supercoiled (sc) pDNA form. (sc) pDNA is unstable and can quickly undergo an irreversible transformation to the open circular (oc) form. This means that the culture supernatant needs to be processed quickly.A purification process that uses high flow rates, has high binding capacities, is scal-able and cGMP compliant must be employed to process the large amounts of plasmids needed. But, does such a process exist?®Plasmid DNA: supercoiled (left drawing), differenttypes (right photo)RNase free process for purifying transfection grade pDNARNA Not detected Genomic DNA Not detected Proteins Not detected by BCA Transformation Successful A260/A280 1.85Endotoxins <0.1EU/mg pDNA Quality of purified plasmid DNA – Biologically Active with impurities under the detection limit Using CIM ®media, the isolation of plasmid DNA no longer requiresthe addition of ribonuclease A (RNase). A 2-step purification solutionis all you need to purify your transfection grade pDNA on any scale.The process employs a CIM ®OH column which captures the pDNAfrom the fermentation pool and separates it from the majority of RNA.This is followed by the polishing step which uses a CIM ®DEAE col-umn (weak anion exchanger). Employing the 8ml columns of thesechemistries, up to 13mg of highly pure pDNA can be isolated.The purified plasmid DNA solution retains its biological activity (con-firmed by bacterial transformation) and complies to the higheststandards of product purity with RNA, proteins, genomic DNA, andendotoxins being under the detection limit.L E M SampleCleared bacterial lysate (E. Coli )Buffer A50mM Tris-HCl, 10mM EDTA, 3.0M (NH 4)2SO 4(pH 7.2)Buffer B50mM Tris-HCl, 10mM EDTA (pH 7.2)Flow rate20ml/min Column CIM ®OH 8ml tube Sample DNA fraction eluted from step 1Buffer A 50mM Tris-HCl, 10mM EDTA (pH 7.2)Buffer B 50mM Tris-HCl, 10mM EDTA, 1.5M NaCl (pH 7.2)Flow rate 20ml/min Column CIM ®DEAE 8ml tube®Purifying Proteins the Simple Waywith Convective Interaction Media®M L F E M L F EM L12M L12kDaWhat is this?Conjoint Liquid ChromatographyConjoint Liquid Chromatography (CLC) is one of the most innova-tive and advantageous features of CIM ®monolithic columns. CLCis the possibility of placing supports with different functional groupsinto one housing-preparing a CLC Monolithic Column. This enablesextremely fast multidimensional chromatography. Now! It is no longer necessary to purchase a large variety of chromatographic columns.Furthermore, there are no extra column effects, such as peak broad-ening, giving much sharper resolution. The idea is even applicable on an industrial scale.CIM ®CLC: Superior performance – Cost saving!with Convective Interaction Media ®Services & Other Formats Upon RequestMajor serviceswith Convective Interaction Media®Joint process development: If you are looking for a partner to help develop a purification process for your pDNA, virus, or pro-tein, BIA Separations is the right choice. BIA Separations and Boehringer Ingelheim co-developed a process for the contract manufacturing of plasmid DNA (pDNA). The manufacturing process produces pDNA for clinical trials and market supply. In addition, BIA Separations is developing small and medium scale kits for the isolation of pDNA on a laboratory scale.Contract Research: BIA Separations contract research laboratory develops and validates analytical methods, performs analysis for pharmacokinetic studies, and isolates and identifies drug impu-rities using HPLC and GC.Designing Industrial Purification Processes: BIA Separations application laboratory utilizes its expertise in liquid chromatog-raphy, monolith and particle based, to design the most efficient, cost effective, and optimized process for biomolecule and tradi-tional small molecule purification.Information and specifications contained herein are, to the best of our knowledge, accurate and represented in good faith. They are intended to help you start working with the new separation technology and are subject to change without notice.BIA Separations shall not be liable for errors contained herein or for incidental or consequentialdamages in connection with the performance or use of CIM®.For more information on our products, visit our home page at: and or contact your local distributor.CIM®technology is covered by US patents 4889632,4923610, 4952349, 5972218, 6319401, 6736973B, 6664305and foreign equivalents. Other patents pending.©2006 by BIA Separations d.o.o. Publication: CPB140508 Printed in Slovenia: 05/2008We reserve the right to alter specification details etc. without prior notice or liability.。


薄膜层析是用作分离和辨识化学物质的层析分析方法中最具多样性的方法之一。
这是一个简单,快速且有效的分离工具,用作量化或质量分析。
身为全球市场的领导者,默克提供您值得信赖的TLC 产品,并拥有广泛的化学性质,尺寸和背景物质可以符合所有您应用的需要。
我们的薄膜板结合了机械性和表面同相性,呈现了不被干扰的分离效能。
供自动化使用的HPTLC 设立了质量控管中可信赖且快速分析的标准。
我们持续增加新的具创新性的产品以符合今日TLC 应用的需求。
为您的分离工作选择最好的TLC 板,进一步了解默克TLC 板如何能让您的实验结果比从前更值得信赖。
高效硅胶薄层层析板(HPTLC)TLC平板自制备用鬆散吸收剂超薄单石型硅平板(UTLC)制备级层析片(PLC)CN-, Diol-, NH2- 修饰硅平板(TLC和HPTLC)氧化铝薄层板(TLC)混合层平板(TLC)纤维素层析板(TLC and HPTLC)Concentrating Zone Plates (TLC, HPTLC和PLC)HPTLC特纯度平板Multiformat Plates (TLC和HPTLC)GLP平板(TLC和HPTLC)配套产品流动相经典硅胶薄层层析板(TLC)RP修饰硅平板(TLC和HPTLC)胜肽分析平板LiChrospher® 球状颗粒HPTLC平板高效硅胶薄层层析板(HPTLC)These HPTLC plates deliver fast and quantitative analysis of complexsamples for manual or automated use. Merck’s HPTLC silica plates wor kthree times faster than conventional TLC plates –and they’re moresensitive. This makes our HPTLC plates perfect for advanced separations.HPTLC and TLC plates use the same type of silica gel 60. But in HPTLC particle sizes range between 4-8 μm, and the mean particle size measures 5-6 μm. This yields a smoother surface and a higher separation power than conventional TLC plates. Highly compact sample bands (thanks to lower band diffusion) and an thin 200 µm layer translate into greatly enhanced sensitivity.目录编号产品105547 HPTLC silica gel 60 ( 高效硅胶层析板) 25 Aluminium sheets 20 x 20 cm105631 HPTLC Silica gel 60 ( 硅胶薄层层析板) 25 Glass plates 10 x 10 cm105641 HPTLC Silica gel 60 ( 高效硅胶层析板) 50 Glass plates 20 x 10 cm105633 HPTLC Silica gel 60 ( 高效硅胶层析板) 100 Glass plates 10 x 10 cm105556 HPTLC Silica gel 60 F254 ( 高效硅胶薄层层析板) 20 Aluminium sheets 5 x 7.5 cm105548 HPTLC Silica gel 60 F254 ( 高效硅胶层析板含荧光指示剂) 25 Aluminium sheets 20 x 20 cmIts small pore size of 100 Å provides larger surface area enabling additional105628 HPTLC Silica gel 60 F254 ( 硅胶薄层层析板含荧光指示剂) 25 Glass plates 10 x 10 cm105642 HPTLC Silica gel 60 F254 ( 硅胶薄层层析板含荧光指示剂) 50 Glass plates 20 x 10 cm105629 HPTLC Silica gel 60 F254 ( 硅胶薄层层析板含荧光指示剂) 100 Glass plates 10 x 10 cm105649 HPTLC silica gel 60 F254 ( HPTLC硅胶60 F254 ) Glass Plates 20 x 10 cm111764 HPTLC Silica gel 60 F254 AMD,extra thin ( 硅胶薄层层析板含荧光指示剂) 25 Glass plates 20 x 10 cm for AMD acc. to DIN 38407-F11115696 HPTLC Silica gel 60 F254s ( 硅胶薄层层析板含荧光指示剂亲水性处理) 25 Glass plates 20 x 10 cm105644 HPTLC Silica gel 60 Multiformat pre-scored to 5 x 5 cm ( 硅胶薄层层析板) 100 Glass plates 10 x 10 cm113749 HPTLC Silica gel 60 with concentrating zone 20 x 2.5 cm ( 高效薄层层析板) 50 Glass plates 20 x 10 cm115552 HPTLC Silica gel 60 WR F254s ( 硅胶薄层层析板含荧光指示剂亲水性处理) 25 Glass plates 20 x 10 cm112363 HPTLC Silica gel 60 WRF254s AMD extra thin ( 硅胶薄层层析板含荧光指示剂亲水性处理) 25 Glass plates 20 x 10 cm for AMD acc. to DIN 38407-F11105616 HPTLC Silica gel F254 ( 硅胶薄层层析板含荧光指示剂) 25 Glass plates 5 x 10 cm超薄单石型硅平板(UTLC)105007 UTLC Silica gel monolithic,10 µm ( 整体化硅胶层析板)制备级层析片(PLC)Preparative thin-layer plates allow users to separate samples that varygreatly in size –from grams down to milligrams. Available with orwithout a fluorescence indicator, PLC plates come as thin as 0.5 cm andas thick as 2 cm. They also use the same proven Merck silica-bindertechnology as in analytical TLC plates.In PLC, samples are typically applied as a band across the entire width of the plate. UV detection is used almost exclusively to render substances visible. To isolate the substance by extraction, users can simply scrape the spot from the layer.目录编号产品105788 PLC Aluminium oxide 60 F254, 1.5 mm ( 制备氧化铝层析板) 12 Glass plates 20 x 20 cm105726 PLC Aluminium oxide 150 F254, 1.5 mm ( 氧化铝制备层析板) 12 Glass plates 20 x 20 cm105744 PLC Silica gel 60 F254, 0.5 mm ( 硅胶薄层层析板含荧光指示剂) 20 Glass plates 20 x 20 cm113895 PLC Silica gel 60 F254, 1 mm ( 制备薄层层析板含荧光指示剂) 15 Glass plates 20 x 20 cm105717 PLC Silica gel 60 F254, 2 mm ( 硅胶薄层层析板含荧光指示剂) 12 Glass plates 20 x 20 cm105637 PLC Silica gel 60 F254+366, 2 mm ( 硅胶薄层层析板含254+366 荧光指示剂2mm ) 12 Glass plates 20 x 20 cm105434 PLC Silica gel 60 RP-18 F254s, 1 mm ( 制备层析板RP-18 含荧光指示剂) 15 Glass plates 20 x 20 cm113894 PLC Silica gel 60, 0.5 mm ( 薄层分析用硅胶片) 20 Glass plates 20 x 20 cm105745 PLC Silica gel 60, 2 mm ( 制备薄层层析板) 12 Glass plates 20 x 20 cmCN-, Diol-, NH2- 修饰硅平板(TLC和HPTLC)These plates tackle extraordinary separation challenges. Our NH2, CN,and diol-modified silica sorbents are less polar then conventional silicaphases, making them ideal for separating hydrophilic or chargedsubstances. Our moderately polar cyano- and diol-modified silica platescan be used for both normal phase and reversed phase systems.An alternative to PEI cellulose, amino-modified NH2 plates provide weak basic ion exchange characteristics with special selectivity for charged compounds. Most modified plates contain F254s, the blue fluorescent, acid stable UV indicator. Fluorescence quenching is used on samples that absorb shortwave UV at 254 nm.目录编号产品116464 HPTLC Silica gel 60 CN F254s ( 硅胶薄层层析板氰基含F254s荧光指示剂) 25 Glass plates 10 x 10 cm112668 HPTLC Silica gel 60 DIOL F254s ( 薄层分析用层析片) 25 Glass plates 10 x 10 cm105636 HPTLC Silica gel 60 DIOL F254s ( Diol薄层层析板含荧光指示剂) 25 Glass plates 20 x 10 cm112572 HPTLC Silica gel 60 NH2 ( 高效薄层层析板) 25 Glass plates 20 x 10 cm113192 HPTLC Silica gel 60 NH2 F254s ( 高效薄层层析板) 25 Glass plates 20 x 10 cm115647 HPTLC Silica gel 60 NH2F254s ( 高效薄层层析板NH2含荧光指示剂) 25 Glass plates 10 x 10 cm105533 TLC Silice gel 60 NH2 F254s ( NH2层析板含荧光指示剂) 20 Aluminium sheets 20 x 20 cm氧化铝薄层板(TLC)These plates are used to analyze basic and neutral compounds atdifferent pH levels.Depending on the pH range in question, professionals can choosebetween two kinds of aluminum oxide plates. Under aqueous conditions,basic aluminum oxide plates are best for separating basic compounds, while neutral plates are ideal for separating neutral compounds.Available with or without a fluorescence indicator, our TLC aluminum plates handle a wide variety of applications thanks to neutral and basic aluminum oxides with 60Å and 150Å pore sizes.目录编号产品105713 TLC Aluminium oxide 60 F254, basic ( 氧化铝薄层层析板含荧光指示剂) 25 Glass plates 20 x 20 cm105731 TLC Aluminium oxide 60 F254, basic ( 氧化铝薄层层析板) 100 Glass plates 5 x 20 cm105550 TLC Aluminium oxide 60 F254, neutral ( 氧化铝60薄层层析板含荧光指示剂中性) 25 Aluminium sheets 20 x 20 cm105581 TLC Aluminium oxide 60 F254, neutral ( 薄层层析用氧化铝片片含荧光指示剂中性) 25 Plastic sheets 20 x 20 cm105551 TLC Aluminium oxide 150 F254, neutral ( 氧化铝150薄层层析板含荧光指示剂中性) 25 Aluminium sheets 20 x 20 cm混合层平板(TLC)Kieselguhr is a natural diatomaceous earth that can used for theseparation of polar or moderately polar substance. Mercks mixed layerplates utilize a combination of classical silica gel 60 and kieselguhrproviding good separation properties for inorganic ions, herbicides andsome steroids.目录编号产品105568 TLC Kieselguhr F254 ( 硅藻土薄层层析板) 25 Aluminium sheets 20 x 20 cm105738 TLC Kieselguhr F254 ( 硅藻土薄层层析板) 25 Glassplates 20 x 20 cm105567 TLC Silica gel 60/kieselguhr F254 ( 硅藻土薄层层析板含荧光指示剂) 25 Aluminium sheets 20 x 20 cm纤维素层析板(TLC and HPTLC)Cellulose plates are used to analyze polar substances. An organicsorbent, cellulose is perfect for separating hydrophilic substances bypartition chromatography. Typical applications include the analysis of amino acids, carbohydrates, and phosphates as well as nucleic acid and nucleic acid derivatives. Merck offers cellulose plates in two grades: TLC for conventional and HPTLC for demanding, high-performance separations.目录编号产品116092 HPTLC Cellulose ( 高效薄层层析板) 25 Aluminium sheets 20 x 20 cm105787 HPTLC Cellulose ( 纤维素薄层层析板) 25 Glass plates 10 x 10 cm105786 HPTLC Cellulose ( 高效纤维素薄层层析板) 50 Glass plates 20 x 10 cm115035 HPTLC Cellulose F ( 高效薄层层析板) 25 Glass plates 10 x 10 cm115036 HPTLC Cellulose F ( 高效薄层层析板) 50 Glass plates 20 x 10 cm105552 TLC Cellulose ( 纤维素薄层层析板) 25 Aluminium sheets 20 x 20 cm105716 TLC Cellulose ( 纤维素薄层层析板) 25 Glass plates 20 x 20 cm105577 TLC Cellulose ( 纤维素薄层层析板) 25 Plastic sheets 20 x 20 cm105730 TLC Cellulose ( 纤维素薄层层析板) 50 Glass plates 10 x 20 cm105632 TLC Cellulose ( 纤维素薄层层析板) 100 Glass plates 10 x 10 cm105574 TLC Cellulose F ( 纤维素薄层层析板) 25 Aluminium sheets 20 x 20 cm105718 TLC Cellulose F ( 纤维素薄层层析板) 25 Glass plates 20 x 20 cm105565 TLC Cellulose F ( 纤维素塑料层析板) 25 Plastic sheets 20 x 20 cm105728 TLC Cellulose F ( 纤维素薄层层析板) 50 Glass plates 10 x 20 cmConcentrating Zone Plates (TLC, HPTLC和PLC)These plates allow users to quickly and easily apply any kind of sample,even large volumes of diluted samples.Merck concentrating zone plates are based on different adsorptionproperties of two adsorbents. The first is a large pore concentratingadsorbent where the samples are applied; the second is a selective layerfor separation. Regardless of the spots’ shape, size, or position, the sample always concentrates as a narrow band where the two adsorbents overlap and where the separation starts.目录编号产品113187 HPTLC Silica gel 60 F254 with concentrating zone 5 x 2.5 cm ( 高效薄层层析板) 25 Glass plates 5 x 10 cm113727 HPTLC Silica gel 60 F254 with concentrating zone 10 x 2.5 cm ( 高效层析硅胶片) 25 Glass plates 10 x 10 cm113728 HPTLC Silica gel 60 F254 with concentrating zone 20 x 2.5 cm ( 高效硅胶层析片) 50 Glass plates 20 x 10 cm115498 HPTLC Silica gel 60 RP-18 F254s with concentrating zone 20 x 2.5 cm ( 薄层层析板RP-18 含荧光指示剂) 25 Glass plates 20 x 10 cm115037 HPTLC Silica gel 60 RP-18 with concentrating zone 20 x 2.5 cm ( 高效薄层层析板) 25 Glass plates 20 x 10 cm for PAH determination acc. to DIN 38409-H13113748 HPTLC Silica gel 60 with concentrating zone 10 x 2.5 cm ( 高级率层析硅胶片) 25 Glass plates 10 x 10 cm113794 PLC Silica gel 60 F254, 0.5 mm with concentrating zone 20 x 4 cm ( 制备层析硅胶片含荧光指示剂) 20 Glass plates 20 x 20 cm 113792 PLC Silica gel 60 F254, 1 mm with concentrating zone 20 x 4 cm ( 制备层析硅胶片含荧光指示剂) 15 Glass plates 20 x 20 cm 113793 PLC Silica gel 60 F254, 2 mm with concentrating zone 20 x 4 cm ( 制备层析硅胶片含荧光指示剂) 12 Glass plates 20 x 20 cm 111846 TLC Silica gel 60 F254 with concentrating zone 10 x 2.5 cm ( 薄层分析用硅胶片) 50 Glass plates 10 x 20 cm105583 TLC Silica gel 60 F254 with concentrating zone 20 x 2.5 cm ( 60 F254s硅胶薄层层析板,浓缩区20 x 2.5 cm ) 25 Aluminium sheets 20 x 20 cm111798 TLC Silica gel 60 F254 with concentrating zone 20 x 2.5 cm ( 硅胶薄层层析板含荧光指示剂) 25 Glass plates 20 x 20 cm111845 TLC Silica gel 60 with concentrating zone 2.5 x 20 cm ( 60硅胶薄层层析板,浓缩区2.5 x 20 cm ) 25 Glass plates 20 x 20 cm 111844 TLC Silica gel 60 with concentrating zone 10 x 2.5 cm ( 薄层分析用硅胶片) 50 Glass plates 10 x 20 cm105582 TLC Silica gel 60 with concentrating zone 20 x 2.5 cm ( 硅胶薄层层析板) 25 Aluminium sheets 20 x 20 cmHPTLC特纯度平板添加到分类最爱Ideal for advanced pharmacopoeia applications, this high-performance plate keeps separations free of contamination. The plate is highly pure and exhibits minimal background, even with middle-polar solvent systems. Its separation performance is an exact match to the related HPTLC plate product. Based on the proven HPTLC 60 F254 plate, the HPTLC premium purity plate comes carefully wrapped in a special, plastic-coated aluminum foil. This prevents plasticizers (such as phthalates) from leaving deposits. Without the foil, the plasticizers could appear as an “unknown extra zone” when mid-polar solvent systems (such as toluene/ethyl acetate (95/5)) are in use, and they could be stained by derivatization reagents (such as anis aldehyde).目录编号产品105648 HPTLC Silica gel 60 F254 Premium Purity ( HPTLC Silica gel 60_F254 Premium Purity ) 50 Glass plates 20 x 10 cm Multiformat Plates (TLC和HPTLC)These pre-scored glass plates fold into smaller formats with just oneeasy snap of the fingers.目录编号产品105635 HPTLC Silica gel 60 F254 Multiformat pre-scored to 5 x 5 cm ( 硅胶高效层析板) ex Glass plates 10 x 10 cm105620 TLC Silica gel 60 F254 Multiformat prescored to 5 x 10 cm ( 硅胶薄层层析板含荧光指示剂含多种规格片) 25 Glass plates 20 x 20 cm,105608 TLC Silica gel 60 F254 Multiformat pre-scored to 5 x 20 cm ( 硅胶薄层层析板含荧光指示剂) 20 Glass plates 20 x 20 cmGLP平板(TLC和HPTLC)These plates feature individual laser coding for GLP applications.Designed for GLP applications, the tops of each of these plates bear anitem, batch, and individual plate number. As a result, lab professionalscan easily record and archive every plate they use. Based on provenMerck silica found in TLC and HPTLC plates, these GLP plates deliverequally unsurpassed separation performance.目录编号产品105564 HPTLC Silica gel 60 F254 GLP ( 硅胶薄层层析板含荧光指示剂GLP ) 25 Glass plates 10 x 10 cm105613 HPTLC Silica gel F254 GLP ( 硅胶薄层层析板含荧光指示剂GLP ) 25 Glass plates 20 x 10 cm105702 TLC Silica gel 60 F254 GLP 25 Glass plates 10 x 20 cm105566 TLC Silica gel 60 F254 GLP ( 硅胶薄层层析板含荧光指示剂GLP ) 25 Glass plates 20 x 20 cm经典硅胶薄层层析板(TLC)Silica gel is the universal adsorbent used in TLC. It allows to carry outalmost every type of separation by suitable choice of the mobile phase.Merck classical silica TLC plates are based on a combination of Mercksilica gel 60 and the addition of a unique polymeric binder resulting in avery adherent and hard surface that will not crack or blister and evenallow writing with a pencil on the surface without risk to damage the layer. The smooth and extremely dense plate surface ensures narrow bands for maximum separation efficiency with lowest background noise e.g. when performing scanning densitometry./classical-silica-tlc-plates-tlc/chinese/c_zWmb.s1LzOQAAAEWuOAfVhTlRP修饰硅平板(TLC和HPTLC)These plates serve two purposes: act as a pilot method for HPLC andallow to choose various solvent system for special separations.RP-modified silica layers from Merck are well suited for many separationchallenges that unmodified silica cannot overcome. These layers useaqueous solvent systems to separate extremely non-polar substances andanalyze particular polar substances that can adapt to ion-pair chromatography.What’s more, RP-modified silica layers are less dependent on atmospheric humidity. Unlike unmodified silica, RP-phases do not exhibit catalytic activity. This makes them the plates of choice for unstable substance that tend to experience oxidative degradation.目录编号产品113726 HPTLC Silica gel 60 RP-2 F254s ( 高效薄层层析板) 25 Glass plates 10 x 10 cm105914 HPTLC Silica gel 60 RP-18 ( 高效RP-18 薄层层析板) 25 Glass plates 20 x 10 cm113724 HPTLC Silica gel 60 RP-18 F254s ( 高级率逆相层析硅胶片) 25 Glass plates 10 x 10 cm114296 HPTLC Silica gel 60 RP-18 W ( 高效薄层层析板) 25 Glass paltes 10 x 20 cm113124 HPTLC Silica gel 60 RP-18 WF254s ( 高效薄层层析板亲水性处理含荧光指示剂) 25 Glass plates 10 x 10 cm113725 HPTLC Silice gel 60 RP-8 F254s ( 高效薄层层析板) 25 Glass plates 10 x 10 cm105746 TLC Silica gel 60 RP-2 ( RP-2 硅胶薄层层析板) 25 Glass plates 20 x 20 cm105747 TLC Silica gel 60 RP-2 F254 (silanized) 25 Glass plates 20*20cm ( TLC硅胶60 RP-2 F254硅烷化25玻板,20*20cm )115684 TLC Silica gel 60 RP-8 F254s ( RP-8薄层层析板含荧光指示剂) 25 Glass plates 5 x 10 cm115388 TLC Silica gel 60 RP-8 F254s ( 薄层层析板RP-8 含荧光指示剂) 25 Glass plates 20 x 20 cm115424 TLC Silica gel 60 RP-8 F254s ( 薄层层析板RP-8 含荧光指示剂) 50 Glass plates 10 x 20 cm105560 TLC Silica gel 60 RP-18 F254s ( 60 RP-18 F254s硅胶薄层层析板) 20 Aluminium sheets 5 x 7.5 cm105559 TLC Silica gel 60 RP-18 F254s ( RP-18薄层层析板含荧光指示剂) 20 Aluminium sheets 20 x 20 cm115685 TLC Silica gel 60 RP-18 F254s ( RP-18薄层层析板含荧光指示剂) 25 Glass plates 5 x 10 cm115389 TLC Silica gel 60 RP-18 F254s ( 薄层层析板RP-18 含荧光指示剂) 25 Glass plates 20 x 20 cm115683 TLC Silica gel 60 RP-18 F254s ( 薄层层析板) 50 Glass plates 5 x 20 cm115423 TLC Silica gel 60 RP-18 F254s ( 薄层层析板RP-18 含荧光指示剂) 50 Glass plates 10 x 20 cm胜肽分析平板At Merck, we’ve enhanced our ProteoChrom® plates to makeseparation highly efficient – especially when peptides and protein digestsneed to be analyzed.ProteoChrom® plates take performance to new levels. Applications arehighly reproducible thanks to optimized separation and staining procedures. With extra-thin 100µm layers, the plates are incredibly sensitive. They’re also perfect for use with aqueous solvent systems since they prove highly stable in water. For added convenience, our ProteoChrom® plates include easy-to-read and detailed protocols.目录编号产品105651 ProteoChrom® HPTLC cellulose plate ( 高效薄层层析板) for peptide analysis 10 x 10 cm105650 ProteoChrom® HPTLC silica gel 60 ( 高效薄层层析板) for peptid analysis 20 x 10 cm105655 ProteoChrom® Peptide Staining Kit ( 多肽染色套组) For 25 stainings of HPTLC platesLiChrospher® 球状颗粒HPTLC平板Unique HPTLC LiChrospher® plates are the first thin layerchromatography plates based on spherical silica particles. They offer theultimate in thin layer chromatography performance and speed enablinghigh throughput analysis of complex samples.HPTLC LiChrospher® plates guarantee:Fast separationsHighly compact spotsLower detection limitsHPTLC LiChrospher® plates are based on Merck proven spherical shaped silica 60 with a rather small particle size of 6-8 µm and narrow particle size distribution of 3-5 µm as it is normally used in HPLC. LiChrospher® posses the very similar broad selectivity as the respective HPTLC plate however plate height, separation numbers and velocity constants are significantly improved.目录编号产品105586 HPTLC LiChrospher® Silica gel 60 F254s ( 硅胶薄层层析板球状颗粒含荧光指示剂) 25 Aluminium sheets 20 x 20 cm115445 HPTLC LiChrospher® Silica gel 60 F254s ( 硅胶薄层层析板Lichrospher 含荧光指示剂) 25 Glass plates 20 x 10 cm105646 HPTLC LiChrospher® Silica gel 60 RP-18 WF254s ( 硅胶薄层层析板Lichrospher RP-18 含荧光指示剂亲水性处理) 25 Glassplates 20 x 10 cm105647 HPTLC LiChrospher® Silica gel 60 WRF254s AMD extra thin ( 硅胶薄层层析板Lichrospher 含荧光指示剂亲水性处理) 25 Glass plates 20 x 10 cm。

semantic analysis exception - column referenceA semantic analysis exception related to a column reference occurs when there is an error in referencing a column in a query. This can happen due to various reasons, such as:1. Misspelling: If the column name is misspelled in the query, the semantic analysis phase may throw an exception as it cannot find the specified column.2. Ambiguity: If there are multiple tables in the query with columns having the same name, the semantic analysis phase may not be able to determine which column is being referred to and throw an exception.3. Missing column: If a column is missing from the table or is renamed, the semantic analysis phase may throw an exception as it cannot find the expected column.To resolve a semantic analysis exception related to a column reference, you can check for the following:1. Verify the spelling of the column name. Make sure it matches the actual column name in the table.2. If there are multiple tables involved, specify the table alias or table name along with the column name to remove any ambiguity.3. Check if the column still exists in the table. If it has been renamed or removed, update the query accordingly.Overall, semantic analysis exceptions related to column references can be resolved by ensuring the correct spelling, addressing any ambiguities, and verifying the existence of the referenced column.。


参考⽂献[1] Meng W., Wei J., Luo X., et al. Separation of β-agonists in pork on a weak cation exchange column by HPLC with fluorescence detection. Analytical Methods,2012, 4(4): 1163.[2] 聂建荣, 朱铭⽴, 连槿, 等. ⾼效液相⾊谱-串联质谱法检测动物尿液中的15 种β-受体激动剂. ⾊谱,2010, 28(8): 759-764.[3] Traynor I., Crooks S., Bowers J., et al. Detection of multi-β-agonist residues in liver matrix by use of a surface plasma resonance biosensor. Analytica Chimica Acta,2003, 483(1): 187-191. [4] Kuiper H., Noordam M., van Dooren-Flipsen M., et al. Illegal use of beta-adrenergic agonists: European Community. Journal of Animal Science,1998, 76(1): 195-207.[5] Watkins L., Jones D., Mowrey D., et al. The effect of various levels of ractopamine hydrochloride on the performance and carcass characteristics of finishing swine. Journal of Animal Science,1990, 68(11): 3588-3595.[6] Parr M. K., Opfermann G., Sch?nzer W. Analytical methods for the detection of clenbuterol. Bioanalysis,2009, 1(2): 437-450.[7] López-Mu?oz F., Alamo C., Rubio G., et al. Half a century since the clinical introduction of chlorpromazine and the birth of modern psychopharmacology. Prog Neuropsychopharmacol Biol Psychiatry,2004, 28(1): 205-208.[8] Goodman L., Gilman A. The pharmacological basis of therapeutics, 7th edn Macmillan. New York,1980: 1054-1105.[9] 王春燕. ⽑细管电泳—电化学发光检测吩噻嗪类药物的研究. 长春理⼯⼤学, 2006.[10] 孙雷, 张骊, 徐倩, et al. 超⾼效液相⾊谱-串联质谱法检测猪⾁和猪肾中残留的10 种镇静剂类药物. ⾊谱,2010, 28(1): 38-42.[11] 顾华兵, 谢洁, 彭涛, et al. 鸡⾁组织中氯丙嗪残留的HPLC-MS/MS 检测⽅法的建⽴. 中国家禽,2014, 36(15): 33-36.[12] Mitchell G., Dunnavan G. Illegal use of beta-adrenergic agonists in the United States. Journal of Animal Science,1998, 76(1): 208-211.[13] Directive C. Council Directive 96/23/EC of 29 April 1996 on measures to monitor certain substances and residues thereof in live animals and animal products and repealing Directives 85/358/EEC and 86/469/EEC and Decisions89/187/EEC and 91/664/EEC. Official Journal L125,1996, 23(5): 10-32.[14] 农业部, 卫⽣部. 禁⽌在饲料和动物饮⽤⽔中使⽤的药物品种⽬录[Z] 农业部公告[2002] 176 号. 2002.[15] Damasceno L., Ventura R., Cardoso J., et al. Diagnostic evidence for the presence of β-agonists using two consecutive derivatization procedures and gas chromatography–mass spectrometric analysis. Journal of Chromatography B,2002,780(1): 61-71.[16] 王培龙. β-受体激动剂及其检测技术研究. 农产品质量与安全,2014, 1): 44-52.[17] Wang L.-Q., Zeng Z.-L., Su Y.-J., et al. Matrix effects in analysis of β-agonists with LC-MS/MS: influence of analyte concentration, sample source, and SPE type. Journal of Agricultural and Food Chemistry,2012, 60(25): 6359-6363.[18] Shao B., Jia X., Zhang J., et al. Multi-residual analysis of 16 β-agonists in pig liver, kidney and muscle by ultra performance liquid chromatography tandem mass spectrometry. Food Chemistry,2009, 114(3): 1115-1121.[19] Josefsson M., Sabanovic A. Sample preparation on polymeric solid phase extraction sorbents for liquid chromatographic-tandem mass spectrometric analysis of human whole blood--a study on a number of beta-agonists and beta-antagonists. Journal of Chromatography A 2006, 1120(1-2):1-12.[20] Zhang Z., Yan H., Cui F., et al. Analysis of Multiple β-Agonist and β-Blocker Residues in Porcine Muscle Using Improved QuEChERS Method and UHPLC-LTQ Orbitrap Mass Spectrometry. Food Analytical Methods,2015: 1-10. [21] Wang P., Liu X., Su X., et al. Sensitive detection of β-agonists in pork tissue with novel molecularly imprinted polymer extraction followed liquid chromatography coupled tandem mass spectrometry detection. Food chemistry,2015, 184(72-79.[22] Li T., Cao J., Li Z., et al. Broad screening and identification of beta-agonists in feed and animal body fluid and tissues using ultra-high performance liquid chromatography-quadrupole-orbitrap high resolution mass spectrometry combined with spectra library search. Food Chem,2016, 192(188-196.[23] Xiong L., Gao Y.-Q., Li W.-H., et al. A method for multiple identification of four β2-Agonists in goat muscle and beefmuscle meats using LC-MS/MS based on deproteinization by adjusting pH and SPE for sample cleanup. Food Science and Biotechnology,2015, 24(5): 1629-1635.[24] Zhang Y., Zhang Z., Sun Y., et al. Development of an Analytical Method for the Determination of β2-Agonist Residues in Animal Tissues by High-Performance Liquid Chromatography with On-line Electrogenerated [Cu (HIO6) 2] 5--Luminol Chemiluminescence Detection. Journal of Agricultural and Food chemistry,2007, 55(13): 4949-4956.[25] Liu W., Zhang L., Wei Z., et al. Analysis of beta-agonists and beta-blockers in urine using hollow fibre-protected liquid-phase microextraction with in situ derivatization followed by gas chromatography/mass spectrometry. Journal of Chromatography A 2009, 1216(28): 5340-5346. [26] Caban M., Mioduszewska K., Stepnowski P., et al. Dimethyl(3,3,3-trifluoropropyl)silyldiethylamine--a new silylating agent for the derivatization of beta-blockers and beta-agonists in environmental samples. Analytica Chimica Acta,2013, 782(75-88.[27] Caban M., Stepnowski P., Kwiatkowski M., et al. Comparison of the Usefulness of SPE Cartridges for the Determination of β-Blockers and β-Agonists (Basic Drugs) in Environmental Aqueous Samples. Journal of Chemistry,2015, 2015([28] Zhang Y., Wang F., Fang L., et al. Rapid determination of ractopamine residues in edible animal products by enzyme-linked immunosorbent assay: development and investigation of matrix effects. J Biomed Biotechnol,2009, 2009(579175.[29] Roda A., Manetta A. C., Piazza F., et al. A rapid and sensitive 384-microtiter wells format chemiluminescent enzyme immunoassay for clenbuterol. Talanta,2000, 52(2): 311-318.[30] Bacigalupo M., Meroni G., Secundo F., et al. Antibodies conjugated with new highly luminescent Eu 3+ and Tb 3+ chelates as markers for time resolved immunoassays. Application to simultaneous determination of clenbuterol and free cortisol in horse urine. Talanta,2009, 80(2): 954-958.[31] He Y., Li X., Tong P., et al. An online field-amplification sample stacking method for the determination of β 2-agonists in human urine by CE-ESI/MS. Talanta,2013, 104(97-102.[32] Li Y., Niu W., Lu J. Sensitive determination of phenothiazines in pharmaceutical preparation and biological fluid by flow injection chemiluminescence method using luminol–KMnO 4 system. Talanta,2007, 71(3): 1124-1129.[33] Saar E., Beyer J., Gerostamoulos D., et al. The analysis of antipsychotic drugs in humanmatrices using LC‐MS (/MS). Drug testing and analysis,2012, 4(6): 376-394.[34] Mallet E., Bounoure F., Skiba M., et al. Pharmacokinetic study of metopimazine by oral route in children. Pharmacol Res Perspect,2015, 3(3): e00130.[35] Thakkar R., Saravaia H., Shah A. Determination of Antipsychotic Drugs Known for Narcotic Action by Ultra Performance Liquid Chromatography. Analytical Chemistry Letters,2015, 5(1): 1-11.[36] Kumazawa T., Hasegawa C., Uchigasaki S., et al. Quantitative determination of phenothiazine derivatives in human plasma using monolithic silica solid-phase extraction tips and gas chromatography–mass spectrometry. Journal of Chromatography A,2011, 1218(18): 2521-2527.[37] Flieger J., Swieboda R. Application of chaotropic effect in reversed-phase liquid chromatography of structurally related phenothiazine and thioxanthene derivatives. J Chromatogr A,2008, 1192(2): 218-224.[38] Tu Y. Y., Hsieh M. M., Chang S. Y. Sensitive detection of piperazinyl phenothiazine drugs by field‐amplified sample stacking in capillary electrophoresis with dispersive liquid–liquid microextraction. Electrophoresis,2015, 36(21-22): 2828-2836.[39] Geiser L., Veuthey J. L. Nonaqueous capillary electrophoresis in pharmaceutical analysis. Electrophoresis,2007, 28(1‐2): 45-57.[40] Lara F. J., García‐Campa?a A. M., Gámiz‐Gracia L., et al. Determination of phenothiazines in pharmaceutical formulations and human urine using capillary electrophoresis with chemiluminescence detection. Electrophoresis,2006,27(12): 2348-2359.[41] Lee H. B., Sarafin K., Peart T. E. Determination of beta-blockers and beta2-agonists in sewage by solid-phase extraction and liquid chromatography-tandem mass spectrometry. J Chromatogr A,2007, 1148(2): 158-167.[42] Meng W., Wei J., Luo X., et al. Separation of β-agonists in pork on a weak cation exchange column by HPLC with fluorescence detection. Analytical Methods,2012, 4(4): 1163-1167. [43] Yang F., Liu Z., Lin Y., et al. Development an UHPLC-MS/MS Method for Detection of β-Agonist Residues in Milk. Food Analytical Methods,2011, 5(1): 138-147.[44] Quintana M., Blanco M., Lacal J., et al. Analysis of promazines in bovine livers by high performance liquid chromatography with ultraviolet and fluorimetric detection. Talanta,2003, 59(2): 417-422.[45] Tanaka E., Nakamura T., Terada M., et al. Simple and simultaneous determination for 12 phenothiazines in human serum by reversed-phase high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci,2007, 854(1-2): 116-120.[46] Kumazawa T., Hasegawa C., Uchigasaki S., et al. Quantitative determination of phenothiazine derivatives in human plasma using monolithic silica solid-phase extraction tips and gas chromatography-mass spectrometry. J ChromatogrA,2011, 1218(18): 2521-2527.[47] Qian J. X., Chen Z. G. A novel electromagnetic induction detector with a coaxial coil for capillary electrophoresis. Chinese Chemical Letters,2012, 23(2): 201-204.[48] Baciu T., Botello I., Borrull F., et al. Capillary electrophoresis and related techniques in the determination of drugs of abuse and their metabolites. TrAC Trends in Analytical Chemistry,2015, 74(89-108.[49] Sirichai S., Khanatharana P. Rapid analysis of clenbuterol, salbutamol, procaterol, and fenoterol in pharmaceuticals and human urine by capillary electrophoresis. Talanta,2008, 76(5):1194-1198.[50] Toussaint B., Palmer M., Chiap P., et al. On‐line coupling of partial filling‐capillary zone electrophoresis with mass spectrometry for the separation of clenbuterol enantiomers. Electrophoresis,2001, 22(7): 1363-1372.[51] Redman E. A., Mellors J. S., Starkey J. A., et al. Characterization of Intact Antibody Drug Conjugate Variants using Microfluidic CE-MS. Analytical chemistry,2016.[52] Ji X., He Z., Ai X., et al. Determination of clenbuterol by capillary electrophoresis immunoassay with chemiluminescence detection. Talanta,2006, 70(2): 353-357.[53] Li L., Du H., Yu H., et al. Application of ionic liquid as additive in determination of three beta-agonists by capillary electrophoresis with amperometric detection. Electrophoresis,2013, 34(2): 277-283.[54] 张维冰. ⽑细管电⾊谱理论基础. 北京:科学出版社,2006.[55] Anurukvorakun O., Suntornsuk W., Suntornsuk L. Factorial design applied to a non-aqueous capillary electrophoresis method for the separation of beta-agonists. J Chromatogr A,2006, 1134(1-2): 326-332.[56] Shi Y., Huang Y., Duan J., et al. Field-amplified on-line sample stacking for separation and determination of cimaterol, clenbuterol and salbutamol using capillary electrophoresis. J Chromatogr A,2006, 1125(1): 124-128.[57] Chevolleau S., Tulliez J. Optimization of the separation of β-agonists by capillary electrophoresis on untreated and C 18 bonded silica capillaries. Journal of Chromatography A,1995, 715(2): 345-354.[58] Wang W., Zhang Y., Wang J., et al. Determination of beta-agonists in pig feed, pig urine and pig liver using capillary electrophoresis with electrochemical detection. Meat Sci,2010, 85(2): 302-305.[59] Lin C. E., Liao W. S., Chen K. H., et al. Influence of pH on electrophoretic behavior of phenothiazines and determination of pKa values by capillary zone electrophoresis. Electrophoresis,2003, 24(18): 3154-3159.[60] Muijselaar P., Claessens H., Cramers C. Determination of structurally related phenothiazines by capillary zone electrophoresis and micellar electrokinetic chromatography. Journal of Chromatography A,1996, 735(1): 395-402.[61] Wang R., Lu X., Xin H., et al. Separation of phenothiazines in aqueous and non-aqueous capillary electrophoresis. Chromatographia,2000, 51(1-2): 29-36.[62] Chen K.-H., Lin C.-E., Liao W.-S., et al. Separation and migration behavior of structurally related phenothiazines in cyclodextrin-modified capillary zone electrophoresis. Journal of Chromatography A,2002, 979(1): 399-408.[63] Lara F. J., Garcia-Campana A. M., Ales-Barrero F., et al. Development and validation of a capillary electrophoresis method for the determination of phenothiazines in human urine in the low nanogram per milliliter concentration range using field-amplified sample injection. Electrophoresis,2005, 26(12): 2418-2429.[64] Lara F. J., Garcia-Campana A. M., Gamiz-Gracia L., et al. Determination of phenothiazines in pharmaceutical formulations and human urine using capillary electrophoresis with chemiluminescence detection. Electrophoresis,2006,27(12): 2348-2359.[65] Yu P. L., Tu Y. Y., Hsieh M. M. Combination of poly(diallyldimethylammonium chloride) and hydroxypropyl-gamma-cyclodextrin for high-speed enantioseparation of phenothiazines bycapillary electrophoresis. Talanta,2015, 131(330-334.[66] Kakiuchi T. Mutual solubility of hydrophobic ionic liquids and water in liquid-liquid two-phase systems for analytical chemistry. Analytical Sciences,2008, 24(10): 1221-1230.[67] 陈志涛. 基于离⼦液体相互作⽤⽑细管电泳新⽅法. 万⽅数据资源系统, 2011.[68] Liu J.-f., Jiang G.-b., J?nsson J. ?. Application of ionic liquids in analytical chemistry. TrAC Trends in Analytical Chemistry,2005, 24(1): 20-27.[69] YauáLi S. F. Electrophoresis of DNA in ionic liquid coated capillary. Analyst,2003, 128(1): 37-41.[70] Kaljurand M. Ionic liquids as electrolytes for nonaqueous capillary electrophoresis. Electrophoresis,2002, 23(426-430.[71] Xu Y., Gao Y., Li T., et al. Highly Efficient Electrochemiluminescence of Functionalized Tris (2, 2′‐bipyridyl) ruthenium (II) and Selective Concentration Enrichment of Its Coreactants. Advanced Functional Materials,2007, 17(6): 1003-1009.[72] Pandey S. Analytical applications of room-temperature ionic liquids: a review of recent efforts. Anal Chim Acta,2006, 556(1): 38-45.[73] Koel M. Ionic Liquids in Chemical Analysis. Critical Reviews in Analytical Chemistry,2005, 35(3): 177-192.[74] Yanes E. G., Gratz S. R., Baldwin M. J., et al. Capillary electrophoretic application of 1-alkyl-3-methylimidazolium-based ionic liquids. Analytical chemistry,2001, 73(16): 3838-3844.[75] Qi S., Cui S., Chen X., et al. Rapid and sensitive determination of anthraquinones in Chinese herb using 1-butyl-3-methylimidazolium-based ionic liquid with β-cyclodextrin as modifier in capillary zone electrophoresis. Journal of Chromatography A,2004, 1059(1-2): 191-198.[76] Jiang T.-F., Gu Y.-L., Liang B., et al. Dynamically coating the capillary with 1-alkyl-3-methylimidazolium-based ionic liquids for separation of basic proteins by capillary electrophoresis. Analytica Chimica Acta,2003, 479(2): 249-254.[77] Jiang T. F., Wang Y. H., Lv Z. H. Dynamic coating of a capillary with room-temperature ionic liquids for the separation of amino acids and acid drugs by capillary electrophoresis. Journal of Analytical Chemistry,2006, 61(11): 1108-1112.[78] Qi S., Cui S., Cheng Y., et al. Rapid separation and determination of aconitine alkaloids in traditional Chinese herbs by capillary electrophoresis using 1-butyl-3-methylimidazoium-based ionic liquid as running electrolyte. Biomed Chromatogr,2006, 20(3): 294-300.[79] Wu X., Wei W., Su Q., et al. Simultaneous separation of basic and acidic proteins using 1-butyl-3-methylimidazolium-based ion liquid as dynamic coating and background electrolyte in capillary electrophoresis. Electrophoresis,2008, 29(11): 2356-2362.[80] Guo X. F., Chen H. Y., Zhou X. H., et al. N-methyl-2-pyrrolidonium methyl sulfonate acidic ionic liquid as a new dynamic coating for separation of basic proteins by capillary electrophoresis. Electrophoresis,2013, 34(24): 3287-3292.[81] Mo H., Zhu L., Xu W. Use of 1-alkyl-3-methylimidazolium-based ionic liquids as background electrolytes in capillary electrophoresis for the analysis of inorganic anions. J Sep Sci,2008, 31(13): 2470-2475.[82] Yu L., Qin W., Li S. F. Y. Ionic liquids as additives for separation of benzoic acid and chlorophenoxy acid herbicides by capillary electrophoresis. Analytica Chimica Acta,2005, 547(2): 165-171.[83] Marszall M. P., Markuszewski M. J., Kaliszan R. Separation of nicotinic acid and itsstructural isomers using 1-ethyl-3-methylimidazolium ionic liquid as a buffer additive by capillary electrophoresis. J Pharm Biomed Anal,2006, 41(1): 329-332.[84] Gao Y., Xu Y., Han B., et al. Sensitive determination of verticine and verticinone in Bulbus Fritillariae by ionic liquid assisted capillary electrophoresis-electrochemiluminescence system. Talanta,2009, 80(2): 448-453.[85] Li J., Han H., Wang Q., et al. Polymeric ionic liquid as a dynamic coating additive for separation of basic proteins by capillary electrophoresis. Anal Chim Acta,2010, 674(2): 243-248.[86] Su H. L., Kao W. C., Lin K. W., et al. 1-Butyl-3-methylimidazolium-based ionic liquids and an anionic surfactant: excellentbackground electrolyte modifiers for the analysis of benzodiazepines through capillary electrophoresis. J ChromatogrA,2010, 1217(17): 2973-2979.[87] Huang L., Lin J. M., Yu L., et al. Improved simultaneous enantioseparation of beta-agonists in CE using beta-CD and ionic liquids. Electrophoresis,2009, 30(6): 1030-1036.[88] Laamanen P. L., Busi S., Lahtinen M., et al. A new ionic liquid dimethyldinonylammonium bromide as a flow modifier for the simultaneous determination of eight carboxylates by capillary electrophoresis. J Chromatogr A,2005, 1095(1-2): 164-171.[89] Yue M.-E., Shi Y.-P. Application of 1-alkyl-3-methylimidazolium-based ionic liquids in separation of bioactive flavonoids by capillary zone electrophoresis. Journal of Separation Science,2006, 29(2): 272-276.[90] Liu C.-Y., Ho Y.-W., Pai Y.-F. Preparation and evaluation of an imidazole-coated capillary column for the electrophoretic separation of aromatic acids. Journal of Chromatography A,2000, 897(1): 383-392.[91] Qin W., Li S. F. An ionic liquid coating for determination of sildenafil and UK‐103,320 in human serum by capillary zone electrophoresis‐ion trap mass spectrometry. Electrophoresis,2002, 23(24): 4110-4116.[92] Qin W., Li S. F. Y. Determination of ammonium and metal ions by capillary electrophoresis–potential gradient detection using ionic liquid as background electrolyte and covalent coating reagent. Journal of Chromatography A,2004, 1048(2): 253-256.[93] Borissova M., Vaher M., Koel M., et al. Capillary zone electrophoresis on chemically bonded imidazolium based salts. J Chromatogr A,2007, 1160(1-2): 320-325.[94] Vaher M., Koel M., Kaljurand M. Non-aqueous capillary electrophoresis in acetonitrile using lonic-liquid buffer electrolytes. Chromatographia,2000, 53(1): S302-S306.[95] Vaher M., Koel M., Kaljurand M. Ionic liquids as electrolytes for nonaqueous capillary electrophoresis. Electrophoresis,2002, 23(3): 426.[96] Vaher M., Koel M. Separation of polyphenolic compounds extracted from plant matrices using capillary electrophoresis. Journal of Chromatography A,2003, 990(1-2): 225-230.[97] Francois Y., Varenne A., Juillerat E., et al. Nonaqueous capillary electrophoretic behavior of 2-aryl propionic acids in the presence of an achiral ionic liquid. A chemometric approach. J Chromatogr A,2007, 1138(1-2): 268-275.[98] Lamoree M., Reinhoud N., Tjaden U., et al. On‐capillary isotachophoresis for loadability enhancement in capillary zone electrophoresis/mass spectrometry of β‐agonists. Biological mass spectrometry,1994, 23(6): 339-345.[99] Huang P., Jin X., Chen Y., et al. Use of a mixed-mode packing and voltage tuning for peptide mixture separation in pressurized capillary electrochromatography with an ion trap storage/reflectron time-of-flight mass spectrometer detector. Analytical chemistry,1999, 71(9):1786-1791.[100] Le D. C., Morin C. J., Beljean M., et al. Electrophoretic separations of twelve phenothiazines and N-demethyl derivatives by using capillary zone electrophoresis and micellar electrokinetic chromatography with non ionic surfactant. Journal of Chromatography A,2005, 1063(1-2): 235-240.。

弱阳离子交换整体柱作为固相萃取材料测定血液中的氟桂利嗪张 骊1 ,杨更亮1,2*,张轶华1,王素敏3,冯莎1(1.河北大学药学院,河北 保定 071002;2.中国科学院化学研究所分子科学中心,北京 100080;3.河北医科大学药理实验室,石家庄 050017)摘要用自制的弱阳离子交换整体柱分析测定血液中的氟桂利嗪,以水做为富集流动相,实现在线富集的同时去除生物样品中蛋白。
考察了该整体柱的性能,方法的回收率及精密度。
实验表明,该整体柱性能良好,再生后可重复使用,具有良好的回收率及精密度。
本方法避免了繁琐的样品预处理,为检测血液中的痕量药物提供了一种简单、经济、快速的新方法。
关键词 弱阳离子交换整体柱;去蛋白;血药浓度;氟桂利嗪基金项目:国家自然科学基金资助项目(No.20675084)和教育部高等学校博士学科点专项科研基金资助项目,教育部优秀青年教师资助计划和中国科学院“百人计划”项目.通讯联系人:杨更亮,男,教授,博士生导师,Tel:(0312)5079788,E-mail:glyang@ .致谢:感谢河北大学附属医院给予本实验的帮助. 引言临床上服用蛋白结合率高的药物后,结合型药物的少量下降都有可能导致游离型药物比例的大量增加,从而导致毒副作用增加,所以临床上监控血液中总药物浓度,特别是游离药物浓度具有重要意义。
体内药物分析主要采用HPLC方法, 但样品预处理比较烦琐,其中的蛋白质严重影响对药物的定量、定性分析。
目前报道的预处理方法包括用液液萃取(LLE)[1-4],固相萃取 (SPE)[5-8]及用有机溶剂或强酸强碱沉淀[9-13]除蛋白质,多步SPE不仅费时费力而且由于使用昂贵的SPE柱成本较高。
近来,整体柱材料由于其高吸附容量、“丰富多彩”的功能修饰方法、制备方法简便和优越的性能引起大家的高度重视。
与传统的填充柱相比,具有高通透性的整体柱可以实现快速分离分析并减少流动相的消耗。
然而,尚未见用整体柱对血液样品中的药物进行分析测定的。

耐火材料用语词典英文耐火材料用语词典。
A B.Abrasive wear: The loss of material caused by friction between hard particles.Acid-resistant material: A material that can resist corrosion caused by acids.Aggregate: A mixture of coarse particles, such as sandor gravel, used in concrete or mortar.Alumina: A chemical compound with the formula Al2O3, used as a refractory material due to its high melting point.Alumina brick: A rectangular unit made from alumina-based refractory material, used in high-temperature applications.Alumina castable: A type of refractory material that can be cast or molded into shape, containing alumina as its main constituent.Alumina refractory: A type of refractory material that has a high alumina content, offering excellent resistance to thermal shock and corrosion.Annealing: A heat treatment process used to relieve internal stresses in a material, improving its mechanical properties.Bauxite: A naturally occurring mineral ore, primarily used as a source of alumina.Bond: The material used to bind refractory particles together, such as clays or cement.Bonded refractory: A type of refractory material that uses a bond to hold the particles together, rather than sintering.Brick: A rectangular unit made from refractory material, used in lining fireboxes, furnaces, and other high-temperature applications.C D.Casting: The process of pouring molten material into a mold to create a desired shape.Castable refractory: A type of refractory material that can be cast or poured into place, offering excellent adaptability and conformability.Cement: A binder used to hold particles together, typically made from limestone and clay.Corrosion: The degradation of a material caused by chemical reactions with its environment.Cracking: The formation of cracks in a material due to thermal stresses or mechanical loads.Dense refractory: A type of refractory material with a high density, offering excellent resistance to heat flux and wear.Ductility: The ability of a material to deform without fracturing under tensile stress.E F.Erosion: The gradual loss of material caused by wear, corrosion, or chemical attack.Expansion joint: A gap or joint designed to allow for thermal expansion and contraction of materials.Firebrick: A rectangular unit made from refractory material, used in high-temperature applications such as furnaces and fireboxes.Fireclay: A type of refractory material with a high silica content, used for high-temperature applications.Flame-resistant material: A material that can resistthe direct impact of flames without significant degradation.Fusion: The process of melting or fusing materials together, typically through the application of heat.Furnace: A device used to heat materials to high temperatures, typically for metallurgical or industrial processes.G H.Graphite: A carbon-based material with high thermal conductivity and resistance to high temperatures.Hardening: The process of increasing a material's hardness and strength through heat treatment or other means.Heat resistance: The ability of a material to withstand high temperatures without significant degradation.High-alumina refractory: A type of refractory material with a high alumina content, offering excellent resistance to thermal shock and wear.Hot face: The inner surface of a refractory lining that is exposed to the hottest temperatures.Hot strength: The ability of a refractory material to maintain its structural integrity at high temperatures.I J.Insulation: Materials used to reduce heat transfer by providing resistance to thermal conduction, convection, and radiation.Integrity: The state of being complete and unbroken; the ability of a material to maintain its structural and functional properties.K L.Lining: The layer or layers of refractory material used to protect the internal surfaces of a furnace or otherhigh-temperature equipment.Low-cement castable: A type of castable refractory that uses a reduced amount of cement as a binder, improving its thermal properties.M N.Masonry: The construction of structures using units such as bricks, blocks, or stones.Melting point: The temperature at which a solid material transforms into a liquid state.Monolithic refractory: A type of refractory material that is poured or gunned into place, forming a continuous, non-unitized lining.Mortar: A material used to bind refractory units together, typically made from sand, lime, and water.O P.Oxidation: The chemical reaction of a material with oxygen, typically resulting in the formation of oxides.Porosity: The presence of voids or pores within a material, affecting its density, strength, and thermal properties.Pyrometallurgy: The branch of metallurgy dealing withthe production of metals through high-temperature processes.Q R.Refractory: A material that can resist hightemperatures without significant degradation or loss of strength.Refractory castable: A type of refractory material that can be cast or molded into shape, offering adaptability and conformability.Refractory cement: A type of cement used in refractory applications, typically with a high alumina or silica content.Refractory gunning mix: A type of monolithic refractory material that is applied by gunning, a process in which the material is shot or pumped into place.Refractory mortar: A type of mortar used in refractory applications, typically with a high alumina or silica content.Resistance to thermal shock: The ability of a material to withstand rapid changes in temperature withoutfracturing or significant degradation.S T.Sintering: The process of joining particles of a material together through heat treatment, typically resulting in increased density and strength.Slag: The solid residue formed during the smelting or refining of ores.Stability: The ability of a material to maintain its physical and chemical properties under varying conditions.Thermal conductivity: The ability of a material to transmit heat through its bulk, measured as the rate of heat flow per unit area per unit temperature gradient.Thermal expansion: The increase in volume or dimensions of a material when heated.Thermal shock resistance: The ability of a material to withstand rapid changes in temperature without sustaining damage.U Z.Unitized refractory: A type of refractory material that consists of preformed units or bricks, which are thenassembled to form a lining.Vitreous: Having a glassy or glassy-like appearance, typically due to high temperatures or fusion processes.Wear resistance: The ability of a material to resist mechanical wear and degradation.Wetting angle: The angle at which a liquid refractory material wets the surface of a solid material, affectingits ability to adhere.Yttrium: A chemical element with the symbol Y, used in certain high-temperature applications due to its excellent thermal properties.Zirconia: A ceramic material with the formula ZrO2, offering excellent resistance to high temperatures and wear.This is a basic dictionary of refractory materials terminology, covering terms related to their properties,composition, and applications. It is not an exhaustive list and may not cover all specialized or niche terms.。

现在所使用的大部分液相柱都是填充柱(packed column),即将所需要的填料先做成球形或无定形的颗粒,然后用高压气泵或是高压液泵将其填充到液相钢管柱中。
这就是许多人常用的液相柱子,商品化的很多,可按不同要求选购。
另一种就是我要说的整体柱。
它的制备就是将预聚合液先引入到柱子当中(钢管柱或玻璃管柱),两端封好,在合适的条件下发生反应,在柱管内形成一个多孔的固态整体,直接将其用在液相柱上。
从该过程中大家可以看出,其制备过程非常简单,省却了许多步骤。
然而其更突出优点是与填充柱相比,它具有更好的通透性。
简单的讲,在填充柱当中,所用填料越细小,得到的柱效会越高,但同时会带来通透变差柱压升高的问题。
因为即便你填上了细小的填料,如果你的流动相流速达不到一定要求的话是体现不出高柱效的。
而整体柱就体现了他的优势,当其骨架为2微米的时候,其通透性相当于直径5微米的填充柱。
在制备的柱子当中,有常规液相上用的和超高压液相上用的两类,主要是柱子直径上的区别。
后者常用毛细管柱。
有必要说一下的是超高压所用的柱子与电色谱(electrochromatography)所用的柱子是相通的,一根柱子做成之后在电色谱中可以用,在超高压液相上也可以用。
电色谱曾被认为是一种非常非常有前途的分离方式,柱效非常高,可达200,000塔板数/米以上,因为该种方法的驱动力是电渗流(electroosmotic flow),不需要压力(但在使用过程中可辅助以压力),它的流动方式是活塞式的,致使在纵向上的扩散减少。
而普通的高效液相,在压力的驱动下流型是抛物线式的,所以其柱效高的也只能达到50,000塔板数/米左右。
但电色谱在做了一段时间后,大家发现了一些比较难以克服的问题,特别是重现性方面的问题,因为电渗流受影响因素较多,较难以严格控制。
另外在使用的过程中经常产生气泡,很烦人。
现在一些仪器公司已经放弃了电色谱仪的研制,甚至有人认为电色谱已经死了(dead)。


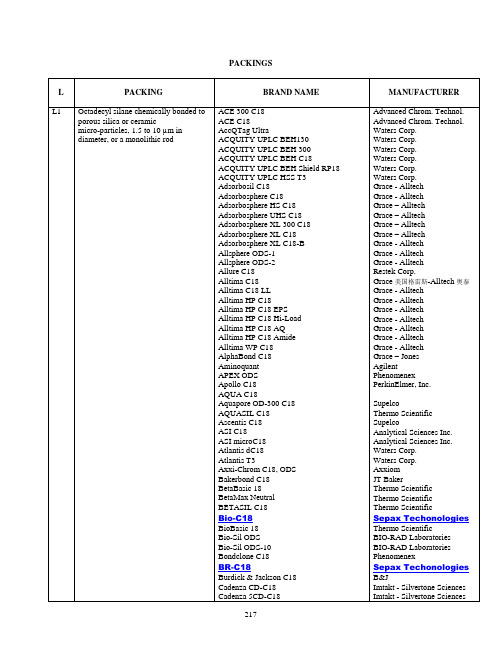
美国药典(USP)中规定的色谱柱类型Chromatographic ReagentsThe following list of packings (L), phases (G), and supports (S) is intended to be a convenient reference for the chromatographer. [note—Particle sizes given in this listing are those generally provided. Where other, usually finer, sizes are required, the individual monograph specifies the desired particle size. Within any category of packings or phases listed below, there may be a wide range of columns available. Where it is necessary to define more specifically the chromatographic conditions, the individual monograph so indicates.]Change to read:PackingsL1—Octadecyl silane chemically bonded to porous silica or ceramic micro-particles, 1.5 to 10 µm in diameter, or a monolithic silica rod.L2—Octadecyl silane chemically bonded to silica gel of a controlled surface porosity that has been bonded to a solid spherical core, 30 to 50 µm in diameter.L3—Porous silica particles, 3 USP31 to 10 µm in diameter, or a monolithic silica rod. USP31L4—Silica gel of controlled surface porosity bonded to a solid spherical core, 30 to 50µm in diameter.L5—Alumina of controlled surface porosity bonded to a solid spherical core, 30 to 50 µm in diameter.L6—Strong cation-exchange packing–sulfonated fluorocarbon polymer coated on a solid spherical core, 30 to 50 µm in diameter.L7—Octylsilane chemically bonded to totally porous silica particles, 1.5 to 10 µm in diameter, or a monolithic silica rod. USP31L8—An essentially monomolecular layer of aminopropylsilane chemically bonded to totally porous silica gel support, 3 to 10 µm in diameter.L9—Irregular or spherical, totally porous silica gel having a chemically bonded, strongly acidic cation-exchange coating, 3 to 10 µm in diameter.L10—Nitrile groups chemically bonded to porous silica particles, 3 to 10 µm in diameter.L11—Phenyl groups chemically bonded to porous silica particles, 1.5 to 10 µm in diameter.L12—A strong anion-exchange packing made by chemically bonding a quaternary amine to a solid silica spherical core, 30 to 50 µm in diameter.L13—Trimethylsilane chemically bonded to porous silica particles, 3 to 10 µm in diameter.L14—Silica gel having a chemically bonded, strongly basic quaternary ammoniumanion-exchange coating, 5 to 10 µm in diameter.L15—Hexylsilane chemically bonded to totally porous silica particles, 3 to 10 µm in diameter.L16—Dimethylsilane chemically bonded to porous silica particles, 5 to 10 µm in diameter.L17—Strong cation-exchange resin consisting of sulfonated cross-linkedstyrene-divinylbenzene copolymer in the hydrogen form, 7 to 11 µm in diameter.L18—Amino and cyano groups chemically bonded to porous silica particles, 3 to 10 µm in diameter.L19—Strong cation-exchange resin consisting of sulfonated cross-linkedstyrene-divinylbenzene copolymer in the calcium form, about 9 µm in diameter.L20—Dihydroxypropane groups chemically bonded to porous silica particles, 5 to 10 µm in diameter.L21—A rigid, spherical styrene-divinylbenzene copolymer, 5 to 10 µm in diameter.L22—A cation-exchange resin made of porous polystyrene gel with sulfonic acid groups, about 10 µm in size.L23—An anion-exchange resin made of porous polymethacrylate or polyacrylate gel with quaternary ammonium groups, about 10 µm in size.L24—A semi-rigid hydrophilic gel consisting of vinyl polymers with numerous hydroxyl groups on the matrix surface, 32 to 63 µm in diameter.[note—Available as YMC-Pack PVA-SIL manufactured by YMC Co., Ltd. and distributed by Waters Corp. ().]L25—Packing having the capacity to separate compounds with a molecular weight range from 100–5000 (as determined by polyethylene oxide), applied to neutral, anionic, and cationicwater-soluble polymers. A polymethacrylate resin base, cross-linked with polyhydroxylated ether (surface contained some residual carboxyl functional groups) was found suitable.L26—Butyl silane chemically bonded to totally porous silica particles, 3 to 10 µm in diameter.L27—Porous silica particles, 30 to 50 µm in diameter.L28—A multifunctional support, which consists of a high purity, 100 , spherical silica substrate that has been bonded with anionic exchanger, amine functionality in addition to a conventional reversed phase C8 functionality.L29—Gamma alumina, reverse-phase, low carbon percentage by weight, alumina-based polybutadiene spherical particles, 5 µm in diameter with a pore volume of 80 .L30—Ethyl silane chemically bonded to totally porous silica particles, 3 to 10 µm in diameter.L31—A hydroxide-selective, strong anion-exchange resin-quaternary amine bonded on latex particles attached to a core of 8.5-µm macroporous particles having a pore size of 2000 and consisting of ethylvinylbenzene cross-linked with 55% divinylbenzene.L32—A chiral ligand-exchange packing–l-proline copper complex covalently bonded to irregularly shaped silica particles, 5 to 10 µm in diameter.L33—Packing having the capacity to separate dextrans by molecular size over a range of4,000 to 500,000 Da. It is spherical, silica-based, and processed to provide pH stability.[note—Available as TSKgel G4000 SWXL from Tosoh Biosep ().]L34—Strong cation-exchange resin consisting of sulfonated cross-linkedstyrene-divinylbenzene copolymer in the lead form, about 9 µm in diameter.L35—A zirconium-stabilized spherical silica packing with a hydrophilic (diol-type) molecular monolayer bonded phase having a pore size of 150 .L36—A 3,5-dinitrobenzoyl derivative of l-phenylglycine covalently bonded to 5-µm aminopropyl silica.L37—Packing having the capacity to separate proteins by molecular size over a range of2,000 to 40,000 Da. It is a polymethacrylate gel.L38—A methacrylate-based size-exclusion packing for water-soluble samples.L39—A hydrophilic polyhydroxymethacrylate gel of totally porous spherical resin.L40—Cellulose tris-3,5-dimethylphenylcarbamate coated porous silica particles, 5 to 20µm in diameter.L41—Immobilized 1-acid glycoprotein on spherical silica particles, 5 µm in diameter.L42—Octylsilane and octadecylsilane groups chemically bonded to porous silica particles, 5 µm in diameter.L43—Pentafluorophenyl groups chemically bonded to silica particles by a propyl spacer, 5 to 10 µm in diameter.L44—A multifunctional support, which consists of a high purity, 60 , spherical silica substrate that has been bonded with a cationic exchanger, sulfonic acid functionality in addition to a conventional reversed phase C8 functionality.L45—Beta cyclodextrin bonded to porous silica particles, 5 to 10 µm in diameter.L46—Polystyrene/divinylbenzene substrate agglomerated with quaternary amine functionalized latex beads, about 10 µm in diameter.L47—High-capacity anion-exchange microporous substrate, fully functionalized with trimethlyamine groups, 8 µm in diameter.[note—Available as CarboPac MA1 and distributed by Dionex Corp. ().]L48—Sulfonated, cross-linked polystyrene with an outer layer of submicron, porous,anion-exchange microbeads, 15 µm in diameter.L49—A reversed-phase packing made by coating a thin layer of polybutadiene onto spherical porous zirconia particles, 3 to 10 µm in diameter.[note—Available as Zirchrom PBD, manufactured by ZirChrom Separations, Inc., distributed by Alltech, .]L50—Multifunction resin with reversed-phase retention and strong anion-exchange functionalities. The resin consists of ethylvinylbenzene, 55% cross-linked with divinylbenzene copolymer, 3 to 15 µm in diameter, and a surface area not less than 350 m2 per g. Substrate is coated with quaternary ammonium functionalized latex particles consisting of styrene cross-linked with divinylbenzene.[note—Available as OmniPac PAX-500 and distributed by Dionex Corp. ().]L51—Amylose tris-3,5-dimethylphenylcarbamate-coated, porous, spherical, silica particles, 5 to 10 µm in diameter.[note—Available as Chiralpak AD from Chiral Technologies, Inc., ().]L52—A strong cation-exchange resin made of porous silica with sulfopropyl groups, 5 to 10µm in diameter.[note—Available as TSK IC SW Cation from Tosoh Biosep ().]L53—Weak cation-exchange resin consisting of ethylvinylbenzene, 55% cross-linked with divinylbenzene copolymer, 3 to 15 µm diameter. Substrate is surface grafted with carboxylic acid and/or phosphoric acid functionalized monomers. Capacity not less than 500 µEq/column.[note—Available as IonPac CS14 distributed by Dionex Corp. ().]L54—A size exclusion medium made of covalent bonding of dextran to highly cross-linked porous agarose beads, about 13 µm in diameter.[note—Available as Superdex Peptide HR 10/30 from Amersham Pharmacia Biotech ().]L55—A strong cation-exchange resin made of porous silica coated with polybutadiene–maleic acid copolymer, about 5 µm in diameter.[note—Available as IC-Pak C M/D from Waters Corp. ().]L56—Propyl silane chemically bonded to totally porous silica particles, 3 to 10 µm in diameter.[note—Available as Zorbax SB-C3 from Agilent Technologies (/chem).]L57—A chiral-recognition protein, ovomucoid, chemically bonded to silica particles, about 5µm in diameter, with a pore size of 120 .[note—Available as Ultron ES-OVM from Agilent Technologies (/chem).]L58—Strong cation-exchange resin consisting of sulfonated cross-linkedstyrene-divinylbenzene copolymer in the sodium form, about 6 to 30 µm 1S (USP31) in diameter.[note—Available as Aminex HPX-87N from Bio-Rad Laboratories, (2000/01 catalog,#125-0143) .]L59—Packing having the capacity to separate proteins by molecular weight over the range of 10 to 500 kDa. It is spherical (10 µm), silica-based, and processed to provide hydrophilic characteristics and pH stability.[note—Available as TSKgel G3000SW Column (analytical column) and TSKgel Guard (guard column) from Tosoh Biosep (part numbers 05789 and 05371, respectively)().]L60—Spherical, porous silica gel, 10 µm or less in diameter, the surface of which has been covalently modified with alkyl amide groups and endcapped.[note—Available as Supelcosil ABZ from Supelco (/supelco).]L61—A hydroxide selective strong anion-exchange resin consisting of a highly cross-linked core of 13 µm microporous particles having a pore size less than 10 units and consisting of ethylvinylbenzene cross-linked with 55% divinylbenzene with a latex coating composed of 85 nm diameter microbeads bonded with alkanol quaternary ammonium ions (6%).[note—Available as Ion Pac AS-11 and AG-11 from Dionex ().]L62—C30 silane bonded phase on a fully porous spherical silica, 3 to 15 µm in diameter.L63— Glycopeptide teicoplanin linked through multiple covalent bonds to a 100- units spherical silica.[Note—Available as Chirobiotic T from Astec ().] 1S (USP31)美国药典(USP)规定的色谱柱编号见下面,是对应的色谱柱类型。

Journal of Chromatography A,1355(2014)228–237Contents lists available at ScienceDirectJournal of ChromatographyAj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c h r o maOne-pot preparation of glutathione–silica hybrid monolith formixed-mode capillary liquid chromatography based on “thiol-ene”click chemistryZian Lin a ,∗,Xiaoqing Tan a ,Ruifang Yu a ,Jiashi Lin b ,Xiaofei Yin c ,Lan Zhang a ,∗,Huanghao Yang aaMinistry of Education Key Laboratory of Analysis and Detection for Food Safety,Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety,College of Chemistry,Fuzhou University,Fuzhou 350116,Fujian,China bCollege of Physical Education,Jimei University,Xiamen 361021,China cThe First Institute of Oceanography,SOA,Qingdao 266061,Chinaa r t i c l ei n f oArticle history:Received 21March 2014Received in revised form 3June 2014Accepted 5June 2014Available online 12June 2014Keywords:Capillary liquid chromatography Organic–inorganic hybrid monolith Glutathione Click chemistry One-potMixed-modea b s t r a c tA novel glutathione (GSH)–silica hybrid monolithic column synthesized via a combination of thiol-ene click reaction and one-pot process was described,where thiol-end GSH organic monomer and 2,2-azobisisobutyronitrile (AIBN)were mixed with hydrolyzed tetramethyloxysilane (TMOS)and ␥-methacryloxypropyltrimethoxysilane (␥-MAPS)and then introduced into a fused-silica capillary for simultaneous polycondensation and “thiol-ene”click reaction to form the GSH–silica hybrid monolith.The effects of the molar ratio of TMOS/␥-MAPS,the amount of GSH,and the volume of porogen on the morphology,permeability and pore properties of the prepared GSH–silica hybrid monoliths were studied in detail.A uniform monolithic network with high porosity was obtained.A series of test com-pounds including alkylbenzenes,amides,and anilines were used to evaluate the retention behaviors of the GSH–silica hybrid monolithic column.The results demonstrated that the prepared GSH–silica hybrid monolith exhibited multiple interactions including hydrophobicity,hydrophilicity,as well as cation exchange interaction.The run-to-run,column-to-column and batch-to-batch reproducibilities of the GSH–silica hybrid monolith for phenols’retention were satisfactory with the relative standard devi-ations (RSDs)less than 1.3%(n =5),2.6%(n =3)and 3.2%(n =3),respectively,indicating the effectiveness and practicability of the proposed method.In addition,the GSH–silica hybrid monolith was applied to the separation of nucleotides,peptides and protein tryptic digests,respectively.The successful applications suggested the potential of the GSH–silica hybrid monolith in complex sample analysis.©2014Elsevier B.V.All rights reserved.1.IntroductionMonolithic materials as stationary phases have been devel-oped as an alternative to the classic particles packing materials for chromatographic separations in high performance liquid chro-matography (HPLC),capillary liquid chromatography (cLC)and capillary electrochromatography (CEC)[1–3],enzyme immobiliza-tion [4]and solid-phase microextraction (SPME)[5]in the past two decades.The most interest in monolithic columns is attributed to∗Corresponding authors at:Fuzhou University,College of Chemistry,Fuzhou,Fujian 350116,China.Tel.:+8659122866135;fax:+8659122866135.E-mail addresses:zianlin@ ,zalin@ (Z.Lin),zlan@ (L.Zhang).their excellent permeability,fast mass transfer kinetics and ease of preparation compared to traditional packed columns [6,7].Based on the nature of the matrix chemistry,monolithic columns can be mainly classified into two types:the organic polymer-based [8–10]and the inorganic silica-based monolithic columns [11–13].Gener-ally speaking,the organic monolithic columns can provide good pH stability and great flexibility to tune the chemical properties of monoliths by using a variety of functional monomers and crosslink-ers [14].However,due to its deficiencies of mechanical stability and desirable porous structure,the organic monolith still has lim-itation in some applications.In contrast,despite the high surface area and high mechanical stability,the surface functionalization of silica-based monolithic columns is labor-intensive and time-consuming.As an alternative,the third type of organic–inorganic hybrid monolithic columns first emerged in 2004[15]and has/10.1016/j.chroma.2014.06.0230021-9673/©2014Elsevier B.V.All rights reserved.Z.Lin et al./J.Chromatogr.A1355(2014)228–237229gained great popularity in recent years since it combines the merits of the organic polymer-based and silica-based monoliths [16–20].In particular,it is noteworthy that Zou’s group[21–23] recently proposed a facile one-pot approach for the synthesis of organic–inorganic hybrid monolithic columns,where the organic functional monomers can be directly incorporated into the inor-ganic silanes,and then polycondensation and polymerization are carried out in one pot by a stepwise reaction temperature.Appar-ently,the utilization of various organic monomers in one-pot process can avoid tedious synthesis of functional silanes,open-ing a new way for obtaining diverse organic–inorganic hybrid monolithic columns with desirable organic functionalities[24–27]. Nevertheless,self-polymerization of functional organic monomers frequently affects the domain size,which may cause bed perme-ability and low column efficiency.Therefore,a facile approach for preparation of the organic–inorganic hybrid monolithic column with appropriate domain size and desirable functional group will highly facilitate the preparation process.Recently,an important segment of“click chemistry”,i.e.radical-based thiol-ene reaction,has been attracting great interest since it possesses several advantages such as simplicity,high effi-ciency,high selectivity and high conversion under mild conditions [28–31].The thiolether linkage formed serves as a strong and sta-ble covalent bond,which is able to withstand harsh conditions. Thiol-ene reaction has been widely employed for the preparation of chromatographic stationary phases including particle-packed columns and polymer-based monoliths[32–36].Besides,silica-based monolithic column with post-modification of hydrophilic n-octadecanethiol via thiol-ene click chemistry has also been reported[37].In these monoliths,however,a limited range of thiol-ene organic monomers was employed with a tradi-tional two-step process.Until recently,Yao’s group[38]and Feng’s group[39]successively developed an one-pot approach for the preparation of organic–inorganic hybrid monoliths via thiol-ene click chemistry,in which tetramethoxysilane(TMOS) and3-mercaptopropyltrimethoxysilane(MPTMS)were adopted as co-precursors and vinyl-containing organic monomers were used as functional moieties.Nevertheless,it is still theoreti-cally unavoidable that the self-polymerization of vinyl-containing organic monomers occurred.Reduced glutathione(GSH),as a hydrophilic tripeptide com-posed of glutamine,cysteine,and glycine,processes a pendant sulfhydryl group and can be reacted with vinyl-containing monomers via“thiol-ene”click chemistry.Furthermore,it contains two free carboxylic acid groups and one amino group,and should exhibit ion-exchange characteristics if tely,a novel type of zwitterionic stationary phase was prepared by covalently bonding GSH on silica gel via click chemistry,which exhibited good hydrophilicity and cation-exchange characteristics[33].However, to the best of our knowledge,no studies on one-pot process in combination with click chemistry for the preparation of GSH–silica hybrid monolithic column have been reported so far.Herein,we reported a facile one-pot approach in combination with“thiol-ene”click reaction for the synthesis of the GSH–silica hybrid monolithic columns by using the hydrolyzed TMOS and␥-methacryloxypropyltrimethoxysilane(␥-MAPS)as co-precursors and GSH as functionalized organic monomer,respectively.The synthetic procedure was as simple and efficient as in situ polymer-ization of polymer-based monolith without any special handling. The influences of the ratio of TMOS to␥-MAPS,the amount of GSH,and the content of porogenic solvent on the morphology, permeability and column performances of the hybrid monoliths were investigated in detail.The applications of the newly designed hybrid separation media to separate a series of small molecules and tryptic digestion of protein were also discussed in this work.2.Experimental2.1.MaterialsTMOS and␥-MAPS were products of Chemical Factory of Wuhan University(Wuhan,China).Ethylene glycol(EG),reduced GSH and poly(ethylene glycol)(PEG,M n=10,000)were purchased from Alfa Aesar(Ward Hill,MA,USA).AIBN was obtained from Tian-jin Chemistry Reagent Factory(Tianjin,China)and recrystallized with methanol(MeOH)prior to use.Sequencing-grade modified trypsin(TPCK-trypsin)was from Promega(Madison,WI,USA). Bovine serum albumin(BSA)was purchased from Beijing Dingguo Co.Ltd(Beijing,China).Five peptides(Tyr-Gly-Gly(YGG),Cys-Tyr-lle-GIn-Asn-Cys-Pro-Leu-Gly(CYIQNCPLG),Tyr-Gly-Gly-Phe-Leu (YGGFL),Arg-Ser-Gly-Phe-Tyr(RSGFY),and His-Cys-Lys-Phe-Trp-Trp(HCKFWW))were purchased from Shanghai Apeptide Co.Ltd (Shanghai,China).Nucleotides including thymidine monophos-phate(TMP),uridine monophosphate(UMP),deoxyadenosine monophosphate(dAMP),guanosine monophosphate(GMP)and cytidine monophosphate(CMP)were obtained from Sigma(St. Louis,MO,USA).Alkylbenzenes,thiourea,anilines,phenols and HPLC-grade acetonitrile(ACN)were obtained from Sinopharm Chemical Reagent(Shanghai,China).All other chemicals were of analytical grade or better.Deionized water was prepared with a Milli-Q water purification system(Millipore,Milford,MA).Capillar-ies with370m o.d.×75m i.d.were the products of Yongnian Optic Fiber Plant(Hebei,China).2.2.InstrumentsAll of hybrid monolithic capillaries with a total length of50cm (effective length25cm)were used unless otherwise stated.All chromatographic experiments were performed on a TriSep-2100 pressurized capillary electrochromatography(pCEC)instrument (this instrument can also be utilized as capillary liquid chro-matography system,Unimicro Technologies,Pleasanton,CA,USA) as described previously[17].Aflow rate of0.05mL/min was used unless otherwise stated and the UV absorbance was monitored at 214nm.Samples were injected through an injection valve with an internal2L sample loop.A four-port splitter was set between the injection valve and the monolithic column to split theflow into a desirable and stableflow rate.Since the splitting ratio was set at400:1,the actual injection volume was about5nL. Scanning electron micrographs(SEM)of the hybrid monolithic col-umn was carried out on a XL-30E scanning electron microscope (Philips,The Netherlands).The adsorption–desorption isotherms of liquid nitrogen were measured by using physisorption analyzer (Micromeritics ASAP2010porosimeter,USA).Fourier transform infrared(FT-IR)spectra of the monolithic materials were recorded using the AVATAR360FT-IR spectrophotometer(Nicolet,Waltham, MA,USA),where3mg powder sample was mixed with100mg KBr.2.3.Preparation of the GSH–silica hybrid monolithic columnIn order to covalently anchor the silica matrix to the capil-lary wall,the inner surface of the capillary was treated with a vinyl silanizing agent according to the previous procedure[40].The schematic preparation of the GSH–silica hybrid monolithic column was illustrated in Fig.1.A prehydrolyzed mixture was prepared by mixing and stirring acetic acid(0.01M,5mL),PEG10000(540mg), TMOS(1.8mL),and␥-MAPS(0.5mL)for1h at ice bath to form a homogeneous solution.Then,30mg of GSH and1wt%AIBN(1mg) dissolved with80L EG were added into0.5mL of the resulting hydrolyzed mixture and then sonicated for20min.Afterward,the mixture was injected into the pretreated capillary to an appropri-ate length with a syringe.When both ends of the capillary were230Z.Lin et al./J.Chromatogr.A 1355(2014)228–237Fig.1.Schematic representation of one-pot synthesis of the GSH–silica hybrid monolithic column via “thiol-ene”click reaction.sealed with two pieces of rubbers,the capillary was incubated at 40◦C for 12h and then increased to 70◦C for another 12h.The obtained GSH–silica hybrid monolithic column was flushed with MeOH to remove the residual monomers and porogens.As a con-trol,a MAPS-silica hybrid monolithic column was also prepared without addition of GSH.2.4.CalculationsColumn permeability (K )reflects through-pore size and external porosity,or a domain size at a constant through-pore size/skeleton size ratio.The permeability of the column was calculated using Darcy’s equation:K =F ×Á×LP × ×r 2(1)where F ,Á,L , P ,and r stand for volume flow rate of the mobile phase,dynamic viscosity of the mobile phase,the col-umn length,the column backpressure,and the inner radius of the column,respectively [41].In this work,MeOH was used as mobile phase and its corresponding value of dynamic viscosity was 0.580×10−3kg/(m s)at 25◦C [42].The retention factor (k )for the analytes was obtained according to the equation,k =(t R −t 0)/t 0,where t R is the retention time of the analytes,and t 0is the retention time of void marker,respectively.2.5.cLC proceduresThe monolithic column was placed in the instrument and equil-ibrated with mobile phase until a stable baseline was obtained.Isocratic elution of a series of small molecules was performed to evaluate the retention behaviors of the GSH–silica hybrid monolithic column in terms of hydrophobic,hydrophilic,and cation-exchange interactions.Different ratio of ACN/H 2O with or without different pH and concentration of phosphate buffered sodium (PBS)were used unless otherwise stated.BSA digestion standard was prepared according to the procedure as described in our previous paper [17]and then separated with a linear gradient elution mode.3.Results and discussion3.1.Preparation and characterization of the GSH–silica hybrid monolithsThe proposed one-pot approach for the preparation of the GSH–silica hybrid monolithic column involves two major pro-cesses:(1)the hydrolysis and condensation of TMOS and ␥-MAPS;(2)the “thiol-ene”click reaction between GSH and vinyl-end silica monolithic matrix.The incorporation of organic monomer during the polycondensation of silanes can effectively inhibit the shrink-age of the silica network [43].Moreover,“thiol-ene”click reaction between the thiol group of GSH and vinyl-end silica monolithic matrix can completely eliminate the self-polymerization of GSH caused by the initiator of AIBN.Since the precondensation composition and reaction tempera-ture have significant impact on the morphology,permeability and separation selectivity of the GSH–silica monolith,several param-eters such as the ratio of TMOS/␥-MAPS,the amount of GSH,the choice of porogen,and reaction temperature were further opti-mized as shown in Table 1.Like the synthesis of other types of polymeric monoliths,the reaction temperature is an important factor in formation of hybrid monolith.In this experiment,the polycondensation was kept con-stant at 40◦C as usually adopted for the preparation of silica-based monoliths based on sol–gel process.On the other hand,the “thiol-ene”click reaction is intensively performed at 60–65◦C as AIBN decomposes between 60◦C and 85◦C to form radicals to initiate “thiol-ene”click reaction [44].Herein,different reaction tempera-tures (60◦C and 70◦C)were investigated and the result showed that little GSH was immobilized by “thiol-ene”click reaction as the reaction temperature was set at 60◦C.In contrast,a dense and homogenous monolithic network with high yield of immobilized GSH was observed when the reaction temperature of 70◦C was applied,which can be confirmed by the following characterizations and chromatographic retention behavior.The selection of porogenic solvent is a crucial factor in for-mation of a homogeneous prepolymerization solution.In view of the hydrophobic AIBN and hydrophilic GSH,some neutral polarTable 1Effect of synthesis parameters on the formation of GSH–silica hybrid monoliths.ColumnTMOS (mL)␥-MAPS (mL)TMOS/␥-MAPSGSH (mg)EG (L)Backpressure (MPa)Permeability (×10−14m 2)a1 1.80.36:1308013.5 2.032 1.80.5 3.6:1308018.61.473 1.80.63:13080>25(too hard to pump)–b4 1.80.5 3.6:1248011.02.495 1.80.53.6:13680Partly dissolved–6 1.80.5 3.6:13060>25(too hard to pump)–7 1.80.5 3.6:13012010.5 2.6081.80.53.6:1801.518.3a Backpressure is obtained with MeOH as the mobile phase at 5L/min without splitting flow;the length of the capillary was kept at 50cm (effective length 25cm).bNo calculation.Z.Lin et al./J.Chromatogr.A1355(2014)228–237231Fig.2.(A and B)SEM images of the GSH–silica hybrid monoliths with different magnifications;(C)The N2isothermal plot with the inset showing the pore-size distribution;(D)FT-IR spectra of the hybrid monolith with and without clicking GSH.solvents(MeOH,ACN,n-propanol,EG and diethylene glycol)were preferred.The results demonstrated that only EG showed good solubility for GSH and AIBN,and a transparent and homogeneous prepolymerization solution could be obtained.In addition,the con-tent of EG was also studied from60L to120L(column2,6 and7in Table1).It was observed that the content of EG less than 80L had great difficulty in dissolving EG and backpressure of the obtained hybrid monolith was very high(>25MPa).In contrast, the volume of EG over80L prolonged the gelation time of pre-condensation,and the obtained monolithic matrix became slack. Therefore,80L of EG wasfinally chosen as the most suitable vol-ume.The ratio of TMOS/␥-MAPS in the reaction mixture affects not only the formation of monolithic network,but also the immobilized amount of GSH via“thiol-ene”click reaction.Herein,the precon-densation solution with the different ratios of TMOS/␥-MAPS from 6:1to3:1was examined(column1–3,Table1).It was observed that the backpressure of the hybrid monoliths gradually increased with the decrease of the ratio of TMOS/␥-MAPS.The results can be explained that the high content of␥-MAPS was prone to the aggre-gation of silica monolithic matrix within the capillary in sol–gel transition,and thus led to the higher backpressure.Although the monolithic columns with low backpressure could be acceptable, the ratio of TMOS/␥-MAPS with3.6:1(column2)was chosen in order to click GSH as more as possible.The amount of GSH in the precondensation mixture affects the permeability and separation selectivity of the hybrid monolith.As observed from Table1(column2,4and5),the higher amount of GSH was,the poorer solubility became.Furthermore,the per-meability of the prepared hybrid monoliths decreased with the increase of GSH added.On the other hand,the high amount of GSH added is considered to be advantageous for improving separation selectivity.Therefore,30mg GSH(in this case,the molar ratio of GSH to␥-MAPS is about1:2,column2)was selected as a favorable compromise with respect to selectivity and permeability.Fig.2(A and B)showed the SEM images of the GSH–silica hybrid monolith at different magnifications.It was observed that a con-tinuous silica monolithic network was obtained on the prepared hybrid monolith and the formed monolithic matrices were attached well to the inner wall of the capillary(Fig.2(A)and the inset). Moreover,a full dense and homogeneous hybrid monolithic matrix with high porosity was obtained(Fig.2(B)),suggesting that the hybrid monolith was stable and no shrinkage occurred during one-pot synthetic procedure.The measured backpressure was linearly (R=0.998)increased from3.6to18.6MPa as theflow rate was increased.This demonstrated that the GSH–silica hybrid mono-lith possessed good mechanical stability under the pressure of 18.6MPa.Accordingly,the permeability of the hybrid monolith was calculated as1.47×10−14m2.Although high back pressure of the hybrid monolith is presented,highflow rate is tolerated due to the silica network and its excellent mechanic stability.In addi-tion,characterization of the pore structure of the GSH–silica hybrid monolith was also performed by nitrogen adsorption–desorption measurement.The specific surface area of the GSH–silica hybrid monolith was calculated to be273.5m2/g with a narrow meso-porous distribution(∼3.6nm,Fig.2(C)),much higher than that232Z.Lin et al./J.Chromatogr.A 1355(2014)228–237Fig.3.(A and B)Relationship between k and ACN concentration on the GSH–silica hybrid monolith (column 2and column 4)and (C)Separation of three solutes with the GSH–silica (column 2)and MAPS-silica hybrid monolith.Conditions:(A):ACN/water;(B):70%ACN;Flow rate (actual flow rate after splitting):0.05mL/min (125nL/min);Pump pressure:4.9MPa;Detection wavelength:214nm;the analytes are (1)toluene;(2)DMF;(3)DMSO.Each of solutes:100ppm.of the MAPS-silica hybrid monolith (26.7m 2/g).Apparently,the high surface area was attributed to the incorporation of GSH via click reaction,which made hybrid monolithic network denser and smaller.Besides,total pore volume and average pore diameter were found to be 0.31cm 3/g,and 4.5nm,better than those of the MAPS-silica hybrid monolith (0.026cm 3/g,and 45nm).Taking together,these results showed that the prepared GSH–silica hybrid mono-lith had a high specific surface area and large pore volume,which made it possible to obtain satisfactory resolution and high column efficiency.FT-IR spectra provide a direct proof of one-pot synthesis of the GSH–silica hybrid monolith (Fig.2(D)).The strong peaks at 1080cm −1and 795cm −1was assigned to the Si O Si vibrations (spectrum a).Characteristic bands of C O and C C stretches at 1720cm −1and 1638cm −1in spectrum a confirmed the existence of ␥-MAPS and the successful polycondensation pared to spectrum a,the peak of 1638cm −1disappeared and some bands of amino groups at 1656cm −1,1465cm −1,and 1356cm −1appeared in spectrum b.Besides,the peaks assigned as COOH (3400cm −1)and –CH 2adsorption (2920cm −1)became stronger.These results confirmed that GSH was successfully immobilized on the surface of silica monolithic matrices after “thiol-ene”click reaction.3.2.Effect of GSH amount on chromatographic selectivity and retention behavior of the GSH–silica hybrid monolithIn order to evaluate and optimize the separation selectiv-ity of the GSH–silica hybrid monolith with different amount of GSH,three solutes (toluene,N ,N -dimethylformamide (DMF)and dimethyl sulfoxide (DMSO))were used as test compounds and a mobile phase containing aqueous/ACN was adopted.The solvent (MeOH)was selected as the void time marker in this system.Fig.3(A and B)presented the retention behaviors of the three solutes on the column 2and column 4with varying ACN content,respectively.Taking column 2as an example,the k value of toluene gradu-ally decreased with the increasing ACN content from 10%to 40%,and closed to zero as the ACN content was over 40%(Fig.3(A)).Furthermore,it was also observed that the hydrophobic toluene was eluted after the polar DMSO and DMF with the ACN content range of 10–40%.The results indicated a typical reversed-phase retention mechanism existed in the GSH–silica hybrid monolith.In contrast,the k values of DMSO and DMF leveled off initially as the ACN content was less than 40%and remarkably increased with the increasing ACN content from 40%to 100%.In this case,the three solutes were eluted in order of increasing hydrophilicity,suggesting a hydrophilic interaction liquid chromatography (HILIC)retention mechanism.Although the similar retention behavior wasobtained with column 4,it was found from Fig.3(B)that the k val-ues of toluene and DMSO in column 4were much lower than those obtained in column 2under the same mobile phase.The results indicated that the column 2has more flexible adjustment in selec-tivity of hydrophobic and hydrophilic interaction than column 4.Fig.3(C)showed the separation of the three solutes with the GSH–silica and MAPS-silica hybrid monoliths.It was observed that absolute baseline separation of toluene,DMF and DMSO can be achieved in the GSH–silica hybrid monolith with the mobile phase of ACN/H 2O (70/30,v/v %).However,almost no retention of the three solutes was observed in the MAPS-silica hybrid monolith despite using the same chromatographic conditions.These results supported the following two conclusions:(1)the successful bond-ing of GSH through “thiol-ene”click reaction;(2)the introduction of GSH responsible for the reversed-phase/HILIC mechanism.The column efficiency of the GSH–silica hybrid monolith was also evaluated under the chromatographic condition as mentioned above.With a flow velocity of 0.47mm/s,the plate heights approx-imate 10.4m for toluene,9.81m for DMF and 8.28m for DMSO were obtained (Fig.S1of Supplementary material)and their cor-responding column efficiencies were ∼96,000plates/m,110,000plates/m and 120,000plates/m.3.3.Hydrophobic interaction chromatography of the GSH–silica hybrid monolithAs mentioned above,the GSH–silica hybrid monolith showed hydrophobic interaction at low ACN content and thus the sep-aration ability of the hybrid monolith was further evaluated by separating five alkylbenzenes.As presented in Fig.4(A),baseline separation of five alkylbenzenes was achieved with the mobile phase of 30%(v/v)ACN in aqueous solution.The five alkylbenzenes were eluted in order of benzene <toluene <ethylbenzene <n -propylbenzene <n -butylbenzene according to increasing hydrophobicity on the GSH–silica hybrid monolith,indicating a typical reversed-phase separation mechanism.Hydrophobic interaction between alkylbenzenes and the GSH–silica hybrid monolith is mainly attributed to the presence of ␥-MAPS.Accord-ingly,the column efficiencies of the five alkylbenzenes were calculated to be 80,000,142,000,160,000,182,000and 124,000plates/m,respectively.In addition,the effect of ACN content on the retention of the five alkylbenzenes was studied (Fig.4(B)),and the result showed that the k values of the five alkylbenzenes decreased with the increase of ACN content,confirming again that the reversed-phase mechanism played a dominant role in the separation of the alkylbenzenes on the GSH–silica hybrid monolith.Z.Lin et al./J.Chromatogr.A1355(2014)228–237233Fig.4.(A)Hydrophobic interaction chromatography for the separation of alkylbenzenes and(B)effect of ACN content on the k values of alkylbenzenes on the GSH–silica hybrid monolith.Conditions for(A):Mobile phase:ACN/water:30/70(v/v%);Flow rate(actualflow rate after splitting):0.05mL/min(125nL/min);Pump pressure:6.7MPa; Detection wavelength:214nm;For(B),all the conditions are same as(A)except for ACN content;(a)the analytes are(1)thiourea(20ppm);(2)benzene(100ppm);(3) toluene(100ppm);(4)ethylbenzene(100ppm);(5)n-propylbenzene(100ppm);(6)n-butylbenzene(100ppm).3.4.Hydrophilic interaction chromatography of the GSH–silica hybrid monolithAs expected,the hydrophilic moieties of the GSH–silica hybrid monolith could be applied to the separation of phenols in HILIC mode(Fig.5).It was observed from Fig.5(A)that the separation of four phenols was achieved with high column efficiencies of70,000–100,000plates/m.the retention order in the GSH–silica hybrid monolith was phenol<catechol<pyrogallol <phloroglucinol.Besides,their corresponding k values increased with the increase of ACN content(Fig.5(B)).Obviously,the HILIC mechanism originated from carboxyl and amino of GSH can respond to the separation of the four phenols based on the obtained results.3.5.Cation-exchange/hydrophobic interaction chromatographyof the GSH–silica hybrid monolithThe GSH–silica hybrid monolith can offer electrostatic interac-tion with charged solutes due to the existence of multiple ionizable moieties(p K1=2.12(COOH),p K2=3.59(COOH),and p K3=8.75 (NH2))on the GSH–silica hybrid monolithic surface.To further investigate the ion-exchange characteristics on the GSH–silica hybrid monolith,the effect of pH values in buffer solution on the retention of charged solutes(2-nitroaniline(p K a=−0.28), o-phenylenediamine(p K a=4.52),1-naphthylamine(p K a=3.92), p-phenylenediamine(p K a=6.04)and benzylamine,p K a=9.33)was conducted in reversed-phase mode and the result was displayed in Fig.6(A).It was observed that only three solutes wereeluted Fig.5.(A)Hydrophilic interaction chromatography for the separation of phenols and(B)effect of ACN content on the k values of phenols on the GSH–silica hybrid monolith. Conditions for(A):Mobile phase:ACN/water:100/0(v/v%);Flow rate(actualflow rate after splitting):0.05mL/min(125nL/min);Pump pressure:3.4MPa;Detection wavelength:214nm;For(B),all the conditions are same as(A)except for ACN content;(a)the analytes are(1)phenol(100ppm);(2)catechol(100ppm);(3)pyrogallol (100ppm);(4)phloroglucinol(100ppm).。


药物分析学报:英文版一、发表说明本团队专注于论文写作与发表服务,擅长案例分析、编程仿真、图表绘制、理论分析等,论文写作、发表300起,具体价格信息联系:本团队并非任何杂志编辑中心,本中心是专业代发论文的机构,与各个杂志社具有长期良好的合作关系,也就是我们通过我们的特殊渠道将论文送给杂志社我们特定的内部人处理论文,保证较高的上稿率,以解决投稿人投稿后焦急的等待与石沉大海的结局。
通过我们可以较为容易达到发表的目地,当然,论文的质量也是重要的基础。
二、期刊简介如下是西安交通大学主办、教育部主管的学术理论刊物,2011年1月由“Academic Journal of Xi’an Jiaotong University:《西安交通大学学报(英文版)》”,经报请教育部、科技部、新闻出版总署批准,于2011年2月更名为“Journal of Pharmaceutical Analysis(JPA,药物分析学报)”。
JPA为国内外公开发行的英文学术期刊,季刊。
JAP主要集中反映与药品质量、用药安全和分析方法等相关的最新研究成果,重点报道:(1)中药(药材、成药、方剂、注射剂)分析;(2)药物复杂体系分析;(3)生物技术药物质量控制技术与方法;(4)药物体内作用过程分析;(5)药物发现过程中的定量与定性分析;(6)分子药理学中的示踪分析;(7)生物药剂学中的定量分析;(8)临床检验与生物分析等。
栏目有:Review、Original Articles 和 Short Communication。
JPA是世界上第一份有关药物分析的专业学术期刊,主编由西安交通大学医学院药物研究所所长、中国药学会药物分析专业委员会主任委员贺浪冲教授担任。
JPA目前被美国《化学文摘》(CA)和荷兰《医学文摘》(EM)收录,并进入中文科技期刊数据库、万方数据库、中国学术光盘数据库(CNKI) 和重庆维普数据库等多家国内著名检索系统。
ReviewPaperQuantitative bioanalytical and analytical method development of dibenzazepine derivative, carbamazepine: A revieworiginalArticle Development and validation of a GC-FID method for quantitative analysis of oleic acid and related fatty acidsORIGlNAL ARTICLEDetermination of cilostazol and its active metabolite 3,4-dehydro cilostazol from small plasma volume by UPLC-MS/MSSHORT COMMUNICATION Capillary electrophoresis to determine entrapment efficiency of a nanostructured lipid carrier loaded with piroxicamINFORMATION Application of analytical instruments in pharmaceutical analysisBioautography and its scope in the field of natural product chemistryORIGINAL ARTICLENon-covalent binding analysis of sulfamethoxazole to human serum albumin:Fluorescence spectroscopy, UV-vis, FT-IR, voltammetric and molecular modelingSHORT COMMUNICATIONIsolation and characterization of a degradation product in leflunomide and a validated selective stability-indicating HPLC-UV method for their quantificationINFORMATIONApplication of analytical instruments in pharmaceutical analysisReviewPaperQuantitative bioanalytical and analytical method development of dibenzazepine derivative, carbamazepine: A revieworiginalArticle Development and validation of a GC-FID method for quantitative analysis of oleic acid and related fatty acidsREVIEW ARTICLEMeasurement uncertainty in pharmaceutical analysis and its applicationORIGINAL ARTICLEMetabolic profiling of plasma from cardiac surgical patients concurrently administered with tranexamic acid:DI-SPME-LC-MS analysisINFORMATIONApplication of analytical instruments in pharmaceutical analysisORIGINAL ARTICLECharge-transfer interaction of drug quinidine with quinol, picric acidand DDQ:Spectroscopic characterization and biological activity studies towards understanding the drug-receptor mechanismINFORMATION Application of analytical instruments in pharmaceutical analysisREVIEW PAPERDevelopment of forced degradation and stability indicating studies of drugs-A review0RIGINAL ARTICLEDirect detection and identification of active pharmaceutical ingredients in intact tablets by helium plasma ionization (HePI) mass spectrometryINFORMATI0NApplication of analytical instruments in pharmaceutical analysisREVIEW PAPERChemometrics: A new scenario in herbal drug standardizationORIGINAL ARTICLEQuantification of anandamide, oleoylethanolamide and palmitoylethanolamide in rodent brain tissue using high performance liquid chromatographyelectrospray mass spectroscopyINFORMATIONApplication of analytical instruments in pharmaceutical analysisREVIEW PAPERPioglitazone:A review of analytical methodsORIGINAL ARTICLERisk evaluation of impurities in topical excipients:The acetol caseSHORT COMMUNICATIONSimultaneous determination of borneol and its metabolite in rat plasma by GC-MS and its application to pharmacokinetic studyINFORMATIONApplication of analytical instruments in pharmaceutical analysisSynthesis of carbon nanosheet from barley and its use as non-enzymatic glucose biosensor0RIGINALARrICLEQuantitation of bivalirudin, a novel anticoagulant peptide, in human plasma by LC-MS/MS: Method development, validation and application to pharmacokineticsLiquid chromatography tandem mass spectrometry method for the estimation of lamotrigine in human plasma:Application to a pharmacokinetic studySimultaneous quantification of prodrug oseltamivir and its metabolite oseltamivir carboxylate in human plasma by LC-MS[MS to support a bioequivalence studyA sensitive, simple and rapid HPLC-MS/MS method for simultaneous quantification of buprenorpine and its N-dealkylated metabolitenorbuprenorphine in human plasmaSPECIALISSUE:HPLCinpharmaceuticalanalysisFused-core particle technology in high-performance liquid chromatography: An overviewREVIEWPAPERApplication of LC–MS/MS for quantitative analysis of glucocorticoids and stimulants in biological fuids0RIGINALARTICLEChiral separation of bavachinin in Fructus Psoraleae and rat plasma by liquid chromatography using permethylated-p-CD as a chiral selectorSHORTCOMMUNICATIONA novel and rapid microbiological assay for ciprofoxacin hydrochlorideORIGINAL ARTICLEPharmacokinetic study of inosiplex tablets in healthy Chinese volunteers by hyphenated HPLC and tandem MS techniquesSHORT COMMUNICATIONI Simultaneous determination of five diterpenoid alkaloids in Herba Delphinii by HPLC/ELSDRapid determination of anti-estrogens by gas chromatography/mass spectrometry in urine:Method validation and application to real samplesImmobilized enzyme reactors in HPLC and its application in inhibitor screening:A reviewSimultaneous determination of pioglitazone and candesartan in human plasma by LC-MS/MS and its application to a human pharmacokinetic studySchisandra chinensis (Turcz.) BaillSimultaneous determination of telmisartan and amlodipine in human plasma by LC-MS]MS and its application in a human pharmacokinetic studyORIGINAL ARTICLESeparation and enrichment of trace ractopamine in biological samples by uniformly-sized molecularly imprinted polymersSHORT COMMUNIC ATION An analytical method for Fe(Ⅱ)and Fe(Ⅲ)determination in pharmaceutical grade iron sucrose complex and sodium ferric gluconate complexORIGINAL ARTICLESIdentification and determination of the major constituents in traditional Chinese medicine Longdan Xiegan Pill by HPLC-DAD-ESI-MSINFORMATIONPublished the papers of GC-MS analysis—Traditional ChinesemedicineRECRUITMENTLecturer/Professor Wanted at Xi'an Jiaotong UniversityREVIEWApplications of HPLC/MS in the analysis of traditional Chinese medicinesORIGlNAL ARTICLESDetermination of phthalate esters in physiological saline solution by monolithic silica spin column extraction methodINFORMATION Published papers about fingerprint and quality control of traditional Chinese medicineRECRUITMENTLecturer/Professor Wanted at Xi'an Jiaotong UniversityORIGINAL ARTICLEHighly sensitive chemiluminescence technology for protein detection using aptamer-based rolling circle amplification platformINFORMATION Published papers about food safetyRECRUITMENTChair Professors and Visiting Professors of "Chang Jiang Scholars Program"Liquid chromatography coupled with time-of-flight and ion trap mass spectrometry for qualitative analysis of herbal medicinesDetection of captopril based on its enhanced resonance lightscattering signals of fluorosurfactant-capped gold nanoparticlesLC-ESI-MS/MS,a modified method for simultaneous quantification of isoflavonoids,flavonoids,alkaloids and saponins in a Chinese herbal preparation Gegen-Qinlian decoctionStudy on chromatographic fingerprint of sarcandra glabra (Thunb.) by microwave-assisted extraction coupled to HPLC/DAD。

New1H-Pyrazole-Containing Polyamine Receptors Able ToComplex L-Glutamate in Water at Physiological pH ValuesCarlos Miranda,†Francisco Escartı´,‡Laurent Lamarque,†Marı´a J.R.Yunta,§Pilar Navarro,*,†Enrique Garcı´a-Espan˜a,*,‡and M.Luisa Jimeno†Contribution from the Instituto de Quı´mica Me´dica,Centro de Quı´mica Orga´nica Manuel Lora Tamayo,CSIC,C/Juan de la Cier V a3,28006Madrid,Spain,Departamento de Quı´mica Inorga´nica,Facultad de Quı´mica,Uni V ersidad de Valencia,c/Doctor Moliner50, 46100Burjassot(Valencia),Spain,and Departamento de Quı´mica Orga´nica,Facultad deQuı´mica,Uni V ersidad Complutense de Madrid,A V plutense s/n,28040Madrid,SpainReceived April16,2003;E-mail:enrique.garcia-es@uv.esAbstract:The interaction of the pyrazole-containing macrocyclic receptors3,6,9,12,13,16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene1[L1],13,26-dibenzyl-3,6,9,12,13,16,-19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene2[L2],3,9,12,13,16,22,-25,26-octaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene3[L3],6,19-dibenzyl-3,6,9,12,13,-16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene4[L4],6,19-diphenethyl-3,6,9,12,13,16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene5[L5],and 6,19-dioctyl-3,6,9,12,13,16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetra-ene6[L6]with L-glutamate in aqueous solution has been studied by potentiometric techniques.The synthesis of receptors3-6[L3-L6]is described for the first time.The potentiometric results show that4[L4]containing benzyl groups in the central nitrogens of the polyamine side chains is the receptor displaying the larger interaction at pH7.4(K eff)2.04×104).The presence of phenethyl5[L5]or octyl groups6[L6]instead of benzyl groups4[L4]in the central nitrogens of the chains produces a drastic decrease in the stability[K eff )3.51×102(5),K eff)3.64×102(6)].The studies show the relevance of the central polyaminic nitrogen in the interaction with glutamate.1[L1]and2[L2]with secondary nitrogens in this position present significantly larger interactions than3[L3],which lacks an amino group in the center of the chains.The NMR and modeling studies suggest the important contribution of hydrogen bonding andπ-cation interaction to adduct formation.IntroductionThe search for the L-glutamate receptor field has been andcontinues to be in a state of almost explosive development.1 L-Glutamate(Glu)is thought to be the predominant excitatory transmitter in the central nervous system(CNS)acting at a rangeof excitatory amino acid receptors.It is well-known that it playsa vital role mediating a great part of the synaptic transmission.2However,there is an increasing amount of experimentalevidence that metabolic defects and glutamatergic abnormalitiescan exacerbate or induce glutamate-mediated excitotoxic damageand consequently neurological disorders.3,4Overactivation ofionotropic(NMDA,AMPA,and Kainate)receptors(iGluRs)by Glu yields an excessive Ca2+influx that produces irreversible loss of neurons of specific areas of the brain.5There is much evidence that these processes induce,at least in part,neuro-degenerative illnesses such as Parkinson,Alzheimer,Huntington, AIDS,dementia,and amyotrophic lateral sclerosis(ALS).6In particular,ALS is one of the neurodegenerative disorders for which there is more evidence that excitotoxicity due to an increase in Glu concentration may contribute to the pathology of the disease.7Memantine,a drug able to antagonize the pathological effects of sustained,but relatively small,increases in extracellular glutamate concentration,has been recently received for the treatment of Alzheimer disease.8However,there is not an effective treatment for ALS.Therefore,the preparation of adequately functionalized synthetic receptors for L-glutamate seems to be an important target in finding new routes for controlling abnormal excitatory processes.However,effective recognition in water of aminocarboxylic acids is not an easy task due to its zwitterionic character at physiological pH values and to the strong competition that it finds in its own solvent.9†Centro de Quı´mica Orga´nica Manuel Lora Tamayo.‡Universidad de Valencia.§Universidad Complutense de Madrid.(1)Jane,D.E.In Medicinal Chemistry into the Millenium;Campbell,M.M.,Blagbrough,I.S.,Eds.;Royal Society of Chemistry:Cambridge,2001;pp67-84.(2)(a)Standaert,D.G.;Young,A.B.In The Pharmacological Basis ofTherapeutics;Hardman,J.G.,Goodman Gilman,A.,Limbird,L.E.,Eds.;McGraw-Hill:New York,1996;Chapter22,p503.(b)Fletcher,E.J.;Loge,D.In An Introduction to Neurotransmission in Health and Disease;Riederer,P.,Kopp,N.,Pearson,J.,Eds.;Oxford University Press:New York,1990;Chapter7,p79.(3)Michaelis,E.K.Prog.Neurobiol.1998,54,369-415.(4)Olney,J.W.Science1969,164,719-721.(5)Green,J.G.;Greenamyre,J.T.Prog.Neurobiol.1996,48,613-63.(6)Bra¨un-Osborne,H.;Egebjerg,J.;Nielsen,E.O.;Madsen,U.;Krogsgaard-Larsen,P.J.Med.Chem.2000,43,2609-2645and references therein.(7)(a)Shaw,P.J.;Ince,P.G.J.Neurol.1997,244(Suppl2),S3-S14.(b)Plaitakis,A.;Fesdjian,C.O.;Shashidharan,S Drugs1996,5,437-456.(8)Frantz,A.;Smith,A.Nat.Re V.Drug Dico V ery2003,2,9.Published on Web12/30/200310.1021/ja035671m CCC:$27.50©2004American Chemical Society J.AM.CHEM.SOC.2004,126,823-8339823There are many types of receptors able to interact with carboxylic acids and amino acids in organic solvents,10-13yielding selective complexation in some instances.However,the number of reported receptors of glutamate in aqueous solution is very scarce.In this sense,one of the few reports concerns an optical sensor based on a Zn(II)complex of a 2,2′:6′,2′′-terpyridine derivative in which L -aspartate and L -glutamate were efficiently bound as axial ligands (K s )104-105M -1)in 50/50water/methanol mixtures.14Among the receptors employed for carboxylic acid recogni-tion,the polyamine macrocycles I -IV in Chart 1are of particular relevance to this work.In a seminal paper,Lehn et al.15showed that saturated polyamines I and II could exert chain-length discrimination between different R ,ω-dicarboxylic acids as a function of the number of methylene groups between the two triamine units of the receptor.Such compounds were also able to interact with a glutamic acid derivative which has the ammonium group protected with an acyl moiety.15,16Compounds III and IV reported by Gotor and Lehn interact in their protonated forms in aqueous solution with protected N -acetyl-L -glutamate and N -acetyl-D -glutamate,showing a higher stability for the interaction with the D -isomer.17In both reports,the interaction with protected N -acetyl-L -glutamate at physiological pH yields constants of ca.3logarithmic units.Recently,we have shown that 1H -pyrazole-containing mac-rocycles present desirable properties for the binding of dopam-ine.18These polyaza macrocycles,apart from having a highpositive charge at neutral pH values,can form hydrogen bonds not only through the ammonium or amine groups but also through the pyrazole nitrogens that can behave as hydrogen bond donors or acceptors.In fact,Elguero et al.19have recently shown the ability of the pyrazole rings to form hydrogen bonds with carboxylic and carboxylate functions.These features can be used to recognize the functionalities of glutamic acid,the carboxylic and/or carboxylate functions and the ammonium group.Apart from this,the introduction of aromatic donor groups appropriately arranged within the macrocyclic framework or appended to it through arms of adequate length may contribute to the recognition event through π-cation interactions with the ammonium group of L -glutamate.π-Cation interactions are a key feature in many enzymatic centers,a classical example being acetylcholine esterase.20The role of such an interaction in abiotic systems was very well illustrated several years ago in a seminal work carried out by Dougherty and Stauffer.21Since then,many other examples have been reported both in biotic and in abiotic systems.22Taking into account all of these considerations,here we report on the ability of receptors 1[L 1]-6[L 6](Chart 2)to interact with L -glutamic acid.These receptors display structures which differ from one another in only one feature,which helps to obtain clear-cut relations between structure and interaction(9)Rebek,J.,Jr.;Askew,B.;Nemeth,D.;Parris,K.J.Am.Chem.Soc.1987,109,2432-2434.(10)Seel,C.;de Mendoza,J.In Comprehensi V e Supramolecular Chemistry ;Vogtle,F.,Ed.;Elsevier Science:New York,1996;Vol.2,p 519.(11)(a)Sessler,J.L.;Sanson,P.I.;Andrievesky,A.;Kral,V.In SupramolecularChemistry of Anions ;Bianchi,A.,Bowman-James,K.,Garcı´a-Espan ˜a,E.,Eds.;John Wiley &Sons:New York,1997;Chapter 10,pp 369-375.(b)Sessler,J.L.;Andrievsky,A.;Kra ´l,V.;Lynch,V.J.Am.Chem.Soc.1997,119,9385-9392.(12)Fitzmaurice,R.J.;Kyne,G.M.;Douheret,D.;Kilburn,J.D.J.Chem.Soc.,Perkin Trans.12002,7,841-864and references therein.(13)Rossi,S.;Kyne,G.M.;Turner,D.L.;Wells,N.J.;Kilburn,J.D.Angew.Chem.,Int.Ed.2002,41,4233-4236.(14)Aı¨t-Haddou,H.;Wiskur,S.L.;Lynch,V.M.;Anslyn,E.V.J.Am.Chem.Soc.2001,123,11296-11297.(15)Hosseini,M.W.;Lehn,J.-M.J.Am.Chem.Soc.1982,104,3525-3527.(16)(a)Hosseini,M.W.;Lehn,J.-M.Hel V .Chim.Acta 1986,69,587-603.(b)Heyer,D.;Lehn,J.-M.Tetrahedron Lett.1986,27,5869-5872.(17)(a)Alfonso,I.;Dietrich,B.;Rebolledo,F.;Gotor,V.;Lehn,J.-M.Hel V .Chim.Acta 2001,84,280-295.(b)Alfonso,I.;Rebolledo,F.;Gotor,V.Chem.-Eur.J.2000,6,3331-3338.(18)Lamarque,L.;Navarro,P.;Miranda,C.;Ara ´n,V.J.;Ochoa,C.;Escartı´,F.;Garcı´a-Espan ˜a,E.;Latorre,J.;Luis,S.V.;Miravet,J.F.J.Am.Chem.Soc .2001,123,10560-10570.(19)Foces-Foces,C.;Echevarria,A.;Jagerovic,N.;Alkorta,I.;Elguero,J.;Langer,U.;Klein,O.;Minguet-Bonvehı´,H.-H.J.Am.Chem.Soc.2001,123,7898-7906.(20)Sussman,J.L.;Harel,M.;Frolow,F.;Oefner,C.;Goldman,A.;Toker,L.;Silman,I.Science 1991,253,872-879.(21)Dougherty,D.A.;Stauffer,D.A.Science 1990,250,1558-1560.(22)(a)Sutcliffe,M.J.;Smeeton,A.H.;Wo,Z.G.;Oswald,R.E.FaradayDiscuss.1998,111,259-272.(b)Kearney,P.C.;Mizoue,L.S.;Kumpf,R.A.;Forman,J.E.;McCurdy,A.;Dougherty,D.A.J.Am.Chem.Soc.1993,115,9907-9919.(c)Bra ¨uner-Osborne,H.;Egebjerg,J.;Nielsen,E.;Madsen,U.;Krogsgaard-Larsen,P.J.Med.Chem.2000,43,2609-2645.(d)Zacharias,N.;Dougherty,D.A.Trends Pharmacol.Sci.2002,23,281-287.(e)Hu,J.;Barbour,L.J.;Gokel,G.W.J.Am.Chem.Soc.2002,124,10940-10941.Chart 1.Some Receptors Employed for Dicarboxylic Acid and N -AcetylglutamateRecognitionChart 2.New 1H -Pyrazole-Containing Polyamine Receptors Able To Complex L -Glutamate inWaterA R T I C L E SMiranda et al.824J.AM.CHEM.SOC.9VOL.126,NO.3,2004strengths.1[L1]and2[L2]differ in the N-benzylation of the pyrazole moiety,and1[L1]and3[L3]differ in the presence in the center of the polyamine side chains of an amino group or of a methylene group.The receptors4[L4]and5[L5]present the central nitrogens of the chain N-functionalized with benzyl or phenethyl groups,and6[L6]has large hydrophobic octyl groups.Results and DiscussionSynthesis of3-6.Macrocycles3-6have been obtained following the procedure previously reported for the preparation of1and2.23The method includes a first dipodal(2+2) condensation of the1H-pyrazol-3,5-dicarbaldehyde7with the corresponding R,ω-diamine,followed by hydrogenation of the resulting Schiff base imine bonds.In the case of receptor3,the Schiff base formed by condensation with1,5-pentanediamine is a stable solid(8,mp208-210°C)which precipitated in68% yield from the reaction mixture.Further reduction with NaBH4 in absolute ethanol gave the expected tetraazamacrocycle3, which after crystallization from toluene was isolated as a pure compound(mp184-186°C).In the cases of receptors4-6, the precursor R,ω-diamines(11a-11c)(Scheme1B)were obtained,by using a procedure previously described for11a.24 This procedure is based on the previous protection of the primary amino groups of1,5-diamino-3-azapentane by treatment with phthalic anhydride,followed by alkylation of the secondary amino group of1,5-diphthalimido-3-azapentane9with benzyl, phenethyl,or octyl bromide.Finally,the phthalimido groups of the N-alkyl substituted intermediates10a-10c were removed by treatment with hydrazine to afford the desired amines11a-11c,which were obtained in moderate yield(54-63%).In contrast with the behavior previously observed in the synthesis of3,in the(2+2)dipodal condensations of7with 3-benzyl-,3-phenethyl-,and3-octyl-substituted3-aza-1,5-pentanediamine11a,11b,and11c,respectively,there was not precipitation of the expected Schiff bases(Scheme1A). Consequently,the reaction mixtures were directly reduced in situ with NaBH4to obtain the desired hexaamines4-6,which after being carefully purified by chromatography afforded purecolorless oils in51%,63%,and31%yield,respectively.The structures of all of these new cyclic polyamines have been established from the analytical and spectroscopic data(MS(ES+), 1H and13C NMR)of both the free ligands3-6and their corresponding hydrochloride salts[3‚4HCl,4‚6HCl,5‚6HCl, and6‚6HCl],which were obtained as stable solids following the same procedure previously reported18for1‚6HCl and2‚6HCl.As usually occurs for3,5-disubstituted1H-pyrazole deriva-tives,either the free ligands3-6or their hydrochlorides show very simple1H and13C NMR spectra,in which signals indicate that,because of the prototropic equilibrium of the pyrazole ring, all of these compounds present average4-fold symmetry on the NMR scale.The quaternary C3and C5carbons appear together,and the pairs of methylene carbons C6,C7,and C8are magnetically equivalent(see Experimental Section).In the13C NMR spectra registered in CDCl3solution, significant differences can be observed between ligand3,without an amino group in the center of the side chain,and the N-substituted ligands4-6.In3,the C3,5signal appears as a broad singlet.However,in4-6,it almost disappears within the baseline of the spectra,and the methylene carbon atoms C6and C8experience a significant broadening.Additionally,a remark-able line-broadening is also observed in the C1′carbon signals belonging to the phenethyl and octyl groups of L5and L6, respectively.All of these data suggest that as the N-substituents located in the middle of the side chains of4-6are larger,the dynamic exchange rate of the pyrazole prototropic equilibrium is gradually lower,probably due to a relation between proto-tropic and conformational equilibria.Acid-Base Behavior.To follow the complexation of L-glutamate(hereafter abbreviated as Glu2-)and its protonated forms(HGlu-,H2Glu,and H3Glu+)by the receptors L1-L6, the acid-base behavior of L-glutamate has to be revisited under the experimental conditions of this work,298K and0.15mol dm-3.The protonation constants obtained,included in the first column of Table1,agree with the literature25and show that the zwitterionic HGlu-species is the only species present in aqueous solution at physiological pH values(Scheme2and Figure S1of Supporting Information).Therefore,receptors for(23)Ara´n,V.J.;Kumar,M.;Molina,J.;Lamarque,L.;Navarro,P.;Garcı´a-Espan˜a,E.;Ramı´rez,J.A.;Luis,S.V.;Escuder,.Chem.1999, 64,6137-6146.(24)(a)Yuen Ng,C.;Motekaitis,R.J.;Martell,A.E.Inorg.Chem.1979,18,2982-2986.(b)Anelli,P.L.;Lunazzi,L.;Montanari,F.;Quici,.Chem.1984,49,4197-4203.Scheme1.Synthesis of the Pyrazole-Containing MacrocyclicReceptorsNew1H-Pyrazole-Containing Polyamine Receptors A R T I C L E SJ.AM.CHEM.SOC.9VOL.126,NO.3,2004825glutamate recognition able to address both the negative charges of the carboxylate groups and the positive charge of ammonium are highly relevant.The protonation constants of L 3-L 6are included in Table 1,together with those we have previously reported for receptors L 1and L 2.23A comparison of the constants of L 4-L 6with those of the nonfunctionalized receptor L 1shows a reduced basicity of the receptors L 4-L 6with tertiary nitrogens at the middle of the polyamine bridges.Such a reduction in basicity prevented the potentiometric detection of the last protonation for these ligands in aqueous solution.A similar reduction in basicity was previously reported for the macrocycle with the N -benzylated pyrazole spacers (L 2).23These diminished basicities are related to the lower probability of the tertiary nitrogens for stabilizing the positive charges through hydrogen bond formation either with adjacent nonprotonated amino groups of the molecule or with water molecules.Also,the increase in the hydrophobicity of these molecules will contribute to their lower basicity.The stepwise basicity constants are relatively high for the first four protonation steps,which is attributable to the fact that these protons can bind to the nitrogen atoms adjacent to the pyrazole groups leaving the central nitrogen free,the electrostatic repulsions between them being therefore of little significance.The remaining protonation steps will occur in the central nitrogen atom,which will produce an important increase in the electrostatic repulsion in the molecule and therefore a reduction in basicity.As stated above,the tertiary nitrogen atoms present in L 4-L 6will also contribute to this diminished basicity.To analyze the interaction with glutamic acid,it is important to know the protonation degree of the ligands at physiological pH values.In Table 2,we have calculated the percentages ofthe different protonated species existing in solution at pH 7.4for receptors L 1-L 6.As can be seen,except for the receptor with the pentamethylenic chains L 3in which the tetraprotonated species prevails,all of the other systems show that the di-and triprotonated species prevail,although to different extents.Interaction with Glutamate.The stepwise constants for the interaction of the receptors L 1-L 6with glutamate are shown in Table 3,and selected distribution diagrams are plotted in Figure 1A -C.All of the studied receptors interact with glutamate forming adduct species with protonation degrees (j )which vary between 8and 0depending on the system (see Table 3).The stepwise constants have been derived from the overall association constants (L +Glu 2-+j H +)H j LGlu (j -2)+,log j )provided by the fitting of the pH-metric titration curves.This takes into account the basicities of the receptors and glutamate (vide supra)and the pH range in which a given species prevails in solution.In this respect,except below pH ca.4and above pH 9,HGlu -can be chosen as the protonated form of glutamate involved in the formation of the different adducts.Below pH 4,the participation of H 2Glu in the equilibria has also to be considered (entries 9and 10in Table 3).For instance,the formation of the H 6LGlu 4+species can proceed through the equilibria HGlu -+H 5L 5+)H 6LGlu 4+(entry 8,Table 3),and H 2Glu +H 4L 4+)H 6LGlu 4(entry 9Table 3),with percentages of participation that depend on pH.One of the effects of the interaction is to render somewhat more basic the receptor,and somewhat more acidic glutamic acid,facilitating the attraction between op-positely charged partners.A first inspection of Table 3and of the diagrams A,B,and C in Figure 1shows that the interaction strengths differ markedly from one system to another depending on the structural features of the receptors involved.L 4is the receptor that presents the highest capacity for interacting with glutamate throughout all of the pH range explored.It must also be remarked that there are not clear-cut trends in the values of the stepwise constants as a function of the protonation degree of the receptors.This suggests that charge -charge attractions do not play the most(25)(a)Martell,E.;Smith,R.M.Critical Stability Constants ;Plenum:NewYork,1975.(b)Motekaitis,R.J.NIST Critically Selected Stability Constants of Metal Complexes Database ;NIST Standard Reference Database,version 4,1997.Table 1.Protonation Constants of Glutamic Acid and Receptors L 1-L 6Determined in NaCl 0.15mol dm -3at 298.1KreactionGluL 1aL 2aL 3bL 4L 5L 6L +H )L H c 9.574(2)d 9.74(2)8.90(3)9.56(1)9.25(3)9.49(4)9.34(5)L H +H )L H 2 4.165(3)8.86(2)8.27(2)8.939(7)8.38(3)8.11(5)8.13(5)L H 2+H )L H 3 2.18(2)7.96(2) 6.62(3)8.02(1) 6.89(5)7.17(6)7.46(7)L H 3+H )L H 4 6.83(2) 5.85(4)7.63(1) 6.32(5) 6.35(6) 5.97(8)L H 4+H )L H 5 4.57(3) 3.37(4) 2.72(8) 2.84(9) 3.23(9)L H 5+H )L H 6 3.18(3) 2.27(6)∑log K H n L41.135.334.233.634.034.1aTaken from ref 23.b These data were previously cited in a short communication (ref 26).c Charges omitted for clarity.d Values in parentheses are the standard deviations in the last significant figure.Scheme 2.L -Glutamate Acid -BaseBehaviorTable 2.Percentages of the Different Protonated Species at pH 7.4H 1L aH 2LH 3LH 4LL 11186417L 21077130L 3083458L 4083458L 51154323L 6842482aCharges omitted for clarity.A R T I C L E SMiranda et al.826J.AM.CHEM.SOC.9VOL.126,NO.3,2004outstanding role and that other forces contribute very importantly to these processes.26However,in systems such as these,which present overlapping equilibria,it is convenient to use conditional constants because they provide a clearer picture of the selectivity trends.27These constants are defined as the quotient between the overall amounts of complexed species and those of free receptor and substrate at a given pH[eq1].In Figure2are presented the logarithms of the effective constants versus pH for all of the studied systems.Receptors L1and L2with a nonfunctionalized secondary amino group in the side chains display opposite trend from all other receptors. While the stability of the L1and L2adducts tends to increase with pH,the other ligands show a decreasing interaction. Additionally,L1and L2present a close interaction over the entire pH range under study.The tetraaminic macrocycle L3is a better(26)Escartı´,F.;Miranda,C.;Lamarque,L.;Latorre,J.;Garcı´a-Espan˜a,E.;Kumar,M.;Ara´n,V.J.;Navarro,mun.2002,9,936-937.(27)(a)Bianchi,A.;Garcı´a-Espan˜a,c.1999,12,1725-1732.(b)Aguilar,J.A.;Celda,B.;Garcı´a-Espan˜a,E.;Luis,S.V.;Martı´nez,M.;Ramı´rez,J.A.;Soriano,C.;Tejero,B.J.Chem.Soc.,Perkin Trans.22000, 7,1323-1328.Table3.Stability Constants for the Interaction of L1-L6with the Different Protonated Forms of Glutamate(Glu) entry reaction a L1L2L3L4L5L6 1Glu+L)Glu L 3.30(2)b 4.11(1)2HGlu+L)HGlu L 3.65(2) 4.11(1) 3.68(2) 3.38(4) 3Glu+H L)HGlu L 3.89(2) 4.48(1) 3.96(2) 3.57(4) 4HGlu+H L)H2Glu L 3.49(2) 3.89(1) 2.37(4) 3.71(2)5HGlu+H2L)H3Glu L 3.44(2) 3.73(1) 2.34(3) 4.14(2) 2.46(4) 2.61(7) 6HGlu+H3L)H4Glu L 3.33(2) 3.56(2) 2.66(3) 4.65(2) 2.74(3) 2.55(7) 7HGlu+H4L)H5Glu L 3.02(2) 3.26(2) 2.58(3) 4.77(2) 2.87(3) 2.91(5) 8HGlu+H5L)H6Glu L 3.11(3) 3.54(2) 6.76(3) 4.96(3) 4.47(3) 9H2Glu+H4L)H6Glu L 2.54(3) 3.05(2) 3.88(2) 5.35(3) 3.66(4) 3.56(3) 10H2Glu+H5L)H7Glu L 2.61(6) 2.73(4) 5.51(3) 3.57(4) 3.22(8) 11H3Glu+H4L)H7Glu L 4.82(2) 4.12(9)a Charges omitted for clarity.b Values in parentheses are standard deviations in the last significantfigure.Figure1.Distribution diagrams for the systems(A)L1-glutamic acid, (B)L4-glutamic acid,and(C)L5-glutamicacid.Figure2.Representation of the variation of K cond(M-1)for the interaction of glutamic acid with(A)L1and L3,(B)L2,L4,L5,and L6.Initial concentrations of glutamate and receptors are10-3mol dm-3.Kcond)∑[(H i L)‚(H j Glu)]/{∑[H i L]∑[H j Glu]}(1)New1H-Pyrazole-Containing Polyamine Receptors A R T I C L E SJ.AM.CHEM.SOC.9VOL.126,NO.3,2004827receptor at acidic pH,but its interaction markedly decreases on raising the pH.These results strongly suggest the implication of the central nitrogens of the lateral polyamine chains in the stabilization of the adducts.Among the N-functionalized receptors,L4presents the largest interaction with glutamate.Interestingly enough,L5,which differs from L4only in having a phenethyl group instead of a benzyl one,presents much lower stability of its adducts.Since the basicity and thereby the protonation states that L4and L5 present with pH are very close,the reason for the larger stability of the L4adducts could reside on a better spatial disposition for formingπ-cation interactions with the ammonium group of the amino acid.In addition,as already pointed out,L4presents the highest affinity for glutamic acid in a wide pH range,being overcome only by L1and L2at pH values over9.This observation again supports the contribution ofπ-cation inter-actions in the system L4-glutamic because at these pH values the ammonium functionality will start to deprotonate(see Scheme2and Figure1B).Table4gathers the percentages of the species existing in equilibria at pH7.4together with the values of the conditional constant at this pH.In correspondence with Figure1A,1C and Figure S2(Supporting Information),it can be seen that for L1, L2,L5,and L6the prevailing species are[H2L‚HGlu]+and[H3L‚HGlu]2+(protonation degrees3and4,respectively),while for L3the main species are[H3L‚HGlu]+and[H4L‚HGlu]2+ (protonation degrees4and5,respectively).The most effective receptor at this pH would be L4which joins hydrogen bonding, charge-charge,andπ-cation contributions for the stabilization of the adducts.To check the selectivity of this receptor,we have also studied its interaction with L-aspartate,which is a competitor of L-glutamate in the biologic receptors.The conditional constant at pH7.4has a value of3.1logarithmic units for the system Asp-L4.Therefore,the selectivity of L4 for glutamate over aspartate(K cond(L4-glu)/K cond(L4-asp))will be of ca.15.It is interesting to remark that the affinity of L4 for zwiterionic L-glutamate at pH7.4is even larger than that displayed by receptors III and IV(Chart1)with the protected dianion N-acetyl-L-glutamate lacking the zwitterionic charac-teristics.Applying eq1and the stability constants reported in ref17,conditional constants at pH7.4of 3.24and 2.96 logarithmic units can be derived for the systems III-L-Glu and IV-L-Glu,respectively.Molecular Modeling Studies.Molecular mechanics-based methods involving docking studies have been used to study the binding orientations and affinities for the complexation of glutamate by L1-L6receptors.The quality of a computer simulation depends on two factors:accuracy of the force field that describes intra-and intermolecular interactions,and an adequate sampling of the conformational and configuration space of the system.28The additive AMBER force field is appropriate for describing the complexation processes of our compounds,as it is one of the best methods29in reproducing H-bonding and stacking stabiliza-tion energies.The experimental data show that at pH7.4,L1-L6exist in different protonation states.So,a theoretical study of the protonation of these ligands was done,including all of the species shown in5%or more abundance in the potentiometric measurements(Table4).In each case,the more favored positions of protons were calculated for mono-,di-,tri-,and tetraprotonated species.Molecular dynamics studies were performed to find the minimum energy conformations with simulated solvent effects.Molecular modeling studies were carried out using the AMBER30method implemented in the Hyperchem6.0pack-age,31modified by the inclusion of appropriate parameters. Where available,the parameters came from analogous ones used in the literature.32All others were developed following Koll-man33and Hopfinger34procedures.The equilibrium bond length and angle values came from experimental values of reasonable reference compounds.All of the compounds were constructed using standard geometry and standard bond lengths.To develop suitable parameters for NH‚‚‚N hydrogen bonding,ab initio calculations at the STO-3G level35were used to calculate atomic charges compatible with the AMBER force field charges,as they gave excellent results,and,at the same time,this method allows the study of aryl-amine interactions.In all cases,full geometry optimizations with the Polak-Ribiere algorithm were carried out,with no restraints.Ions are separated far away and well solvated in water due to the fact that water has a high dielectric constant and hydrogen bond network.Consequently,there is no need to use counteri-ons36in the modelization studies.In the absence of explicit solvent molecules,a distance-dependent dielectric factor quali-tatively simulates the presence of water,as it takes into account the fact that the intermolecular electrostatic interactions should vanish more rapidly with distance than in the gas phase.The same results can be obtained using a constant dielectric factor greater than1.We have chosen to use a distance-dependent dielectric constant( )4R ij)as this was the method used by Weiner et al.37to develop the AMBER force field.Table8 shows the theoretical differences in protonation energy(∆E p) of mono-,bi-,and triprotonated hexaamine ligands,for the (28)Urban,J.J.;Cronin,C.W.;Roberts,R.R.;Famini,G.R.J.Am.Chem.Soc.1997,119,12292-12299.(29)Hobza,P.;Kabelac,M.;Sponer,J.;Mejzlik,P.;Vondrasek,put.Chem.1997,18,1136-1150.(30)Cornell,W.D.;Cieplak,P.;Bayly,C.I.;Gould,I.R.;Merz,K.M.,Jr.;Ferguson,D.M.;Spelmeyer,D.C.;Fox,T.;Caldwell,J.W.;Kollman,P.A.J.Am.Chem.Soc.1995,117,5179-5197.(31)Hyperchem6.0(Hypercube Inc.).(32)(a)Fox,T.;Scanlan,T.S.;Kollman,P.A.J.Am.Chem.Soc.1997,119,11571-11577.(b)Grootenhuis,P.D.;Kollman,P.A.J.Am.Chem.Soc.1989,111,2152-2158.(c)Moyna,G.;Hernandez,G.;Williams,H.J.;Nachman,R.J.;Scott,put.Sci.1997,37,951-956.(d)Boden,C.D.J.;Patenden,put.-Aided Mol.Des.1999, 13,153-166.(33)/amber.(34)Hopfinger,A.J.;Pearlstein,put.Chem.1984,5,486-499.(35)Glennon,T.M.;Zheng,Y.-J.;Le Grand,S.M.;Shutzberg,B.A.;Merz,K.M.,put.Chem.1994,15,1019-1040.(36)Wang,J.;Kollman,P.A.J.Am.Chem.Soc.1998,120,11106-11114.Table4.Percentages of the Different Protonated Adducts[HGlu‚H j L](j-1)+,Overall Percentages of Complexation,andConditional Constants(K Cond)at pH7.4for the Interaction ofGlutamate(HGlu-)with Receptors L1-L6at Physiological pH[H n L‚HGlu]an)1n)2n)3n)4∑{[H n L‚HGlu]}K cond(M-1)L13272353 2.44×103L2947763 4.12×103L31101324 3.99×102L423737581 2.04×104L51010222 3.51×102L6121224 3.64×102a Charges omitted for clarity.A R T I C L E S Miranda et al. 828J.AM.CHEM.SOC.9VOL.126,NO.3,2004。

Journal of Chromatography A,1217(2010)858–880Contents lists available at ScienceDirectJournal of ChromatographyAj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c h r o maReviewThe challenges of the analysis of basic compounds by high performance liquid chromatography:Some possible approaches for improved separationsDavid V.McCalley ∗Centre for Research in Biomedicine,University of the West of England,Frenchay,Bristol BS161QY,UKa r t i c l e i n f o Article history:Available online 3December 2009Keywords:HPLCBasic compounds Stationary phases Reversed-phase HILICa b s t r a c tThis review considers some of the difficulties encountered with the analysis of ionised bases using reversed-phase chromatography,such as detrimental interaction with column silanol groups,and over-loading which both lead to poor peak shapes.Methods of overcoming these problems in reversed-phase (RP)separations,by judicious selection of the column and mobile phase conditions,are discussed.Hydrophilic interaction chromatography is considered as an alternative method for the separation of some basic compounds.©2009Elsevier B.V.All rights reserved.Contents 1.Introduction.........................................................................................................................................8592.Choice of column....................................................................................................................................8592.1.Column testing procedures..................................................................................................................8592.2.The Tanaka test and the Snyder hydrophobic subtraction parison of results with direct peak shape measurements...8602.3.Monolithic silica columns ...................................................................................................................8632.4.Slow column equilibration.Anion-exchange behaviour of alkylsilica RP columns e of column materials other than silica...................................................................................................8653.Choice of mobile phase..............................................................................................................................8663.1.Choice of modifier ...........................................................................................................................8663.2.Choice of mobile phase pH.Problem of reduced retention of bases at low pH.............................................................8664.Overloading .........................................................................................................................................8674.1.Overview of the problem....................................................................................................................8674.2.Possible causes of overloading ..............................................................................................................8684.3.Effect of buffer anion on overload...........................................................................................................8704.4.Overloading on mixed-mode reversed-phase/cation-exchange columns..................................................................8714.5.Effect of buffer pH on overloading ..........................................................................................................8715.Temperature effects.................................................................................................................................8736.Hydrophilic interaction chromatography (HILIC)..................................................................................................8747.Concluding remarks.................................................................................................................................8787.1.Overloading..................................................................................................................................8787.2.Selection of mobile phase pH................................................................................................................8787.3.Quality and choice of column ...............................................................................................................8797.4.Temperature.................................................................................................................................8797.5.Alternative separation mechanisms—e.g.HILIC.............................................................................................879References...........................................................................................................................................879DOI of original article:10.1016/j.chroma.2009.11.067.∗Tel.:+441173282469;fax:+441173282904.E-mail address:david.mccalley@0021-9673/$–see front matter ©2009Elsevier B.V.All rights reserved.doi:10.1016/j.chroma.2009.11.068D.V.McCalley/J.Chromatogr.A1217(2010)858–8808591.IntroductionThe analysis of basic compounds by high performance liquid chromatography(HPLC)continues to be of interest,as over70% of pharmaceuticals are bases(with about20%being acids)[1–3].A large number of compounds of biomedical and biological signifi-cance are also bases.Reversed-phase(RP)separations are by far the most common in liquid chromatography(LC),due to advantages that include ease of use with gradient elution,compatibility with aqueous samples,versatility of the retention mechanism allowing changes in the separation to be brought about by changes in pH, organic modifier or additives,and long experience with the tech-nique,allowing the rapid establishment of suitable experimental conditions for the analysis of a given sample[4].Nevertheless,it has been recognised for a long time that the analysis of basic com-pounds poses particular difficulties in RP separations.Many of these problems are associated with the complex structure of the surface in silica-based RP packings,shown in Fig.1.The surface concentra-tion of silanols on bare silica is reported to be about8.0mol m−2 [5].C18ligands are too bulky to react completely with all silanols; thus,a maximum coverage of4–4.5mol m−2can be achieved.A further number of reactive silanols can be“endcapped”by reac-tion with smaller silylating agents such as trimethylchlorosilane, but as many as50%of the original silanol groups remain unreacted on a typical RP column.The average p K a of these silanol groups is around7.1,but their acidity can be enhanced by the presence of metal impurities in the silica.Some groups appear to be suffi-ciently acidic that their ionisation cannot be entirely suppressed using acidic mobile phases with a pH within the stability limit of typical RP columns(2.5–7.5).Over this range of operational pH values,basic compounds are likely to be ionised,leading to ionic interactions with ionised silanol groups.BH++SiO−M+→SiO−BH++M+(1) where BH+represents the protonated base,and M+the mobile phase buffer cation.The problem of poor column efficiency(N)and exponentially tailing peaks shown by small quantities of bases is often attributed to this mixed mechanism process of hydropho-bic interaction and ion-exchange with the silanols.The slower sorption–desorption kinetics of silanol ion-exchange sites(kinetic tailing)with sample ions may be responsible[6],which will occur regardless of sample size.The simple existence of two retention processes cannot per se be the sole cause of tailing,as mixed-mode phases with carboxylic acid functions embedded within a hydrophobic chain can show excellent peak symmetry for bases[7]. However,the kinetics of interaction of such embedded groups,and the stereochemistry around the active site,could be completely dif-ferent from that of ionised silanols,which may be buried beneath the hydrophobic chains on classical C18phases.Instead of sim-ple ion-exchange sites,Neue et al.[8]have proposed the existence of strong synergistic sites with combined RP and ion-exchange properties.The overall retention for bases was described by the equation:k=k RP+k IX+k∗RP k∗IX(2) where k is the total retention,k RP is the hydrophobic contribu-tion,k IX is the ion-exchange contribution from surface silanols,and k∗RP k∗IX is a multiplicative contribution of both processes.These syn-ergistic sites could correspond to the subset of very high-energy sites with slow kinetics which have been long suspected to be the cause of exponential tailing for bases,as they appear to be domi-nant in the retention process.It was shown that this type of tailing is not responsive to small changes in sample load in RP–LC at low pH[6].This result might indicate that exponential tailing is not caused by overload of a small number of strong sites on the column. In contrast,overload often gives rise to right-angled triangle peak shapes when ionisation of silanols is suppressed in RP–LC when working at low pH.Overload tailing still occurs even for the most modern columns operated under conditions where there are no or a negligible number of ionised silanols on the column surface.It was recognised more than20years ago that bonded phases synthesised from pure silica(Type B phases)made from the hydrol-ysis of metal-free tetraalkoxysilanes resulted in reduced silanol acidity,and their use has considerably improved the analysis of bases[9].Only small contamination of such materials occurs dur-ing the processing of such packings,or from the water used in the hydrolysis.Nevertheless,some other features of the analysis of these solutes(such as overloading)remain problematic,and these issues have not been resolved by the use of high-purity silica.Already in1988,Snyder and co-workers[10]had reviewed the problems of analysis of basic solutes and had proposed some pos-sible solutions.The following recommendations were made: (a)Judicious selection of the column to reduce the number of avail-able acidic sites.(b)Reduction of the mobile phase pH to suppress ionisation of thesilanols.(c)Increasing the mobile phase pH above the analyte p K a,such thatthe analyte is unprotonated.(d)Addition of a silanol blocker such as triethylamine to the mobilephase to interact preferentially with ionised silanols.(e)Reduction of the sample concentration to alleviate the satura-tion of the acidic sites.Most of the arguments in this paper remain true more than20 years later,and these conclusions can be used as a simple guide for the chromatographer aiming to achieve the best separations for basic solutes.Perhaps only the use of silanol blocking agents has fallen somewhat out of favour,as these are less necessary with modern high-purity silica phases,and can also have some undesirable effects.Such effects include the generation of addi-tional background in HPLC–MS,the difficulty of removal from the stationary phase after use leading to permanent alteration of its properties,and even chemical reaction with some solute types.This topic,and some other well-known aspects of the chromatography of bases have been covered adequately in earlier reviews[11–13]. However,other features of the chromatography of these“difficult”compounds are still extensively debated in the literature,for exam-ple,the problem of their ready overloading in RP separations.This review will concentrate on the latest research in these topics,while attempting to summarise briefly previousfindings.Thus,it will con-sider RP column choice by use of evaluation data obtained from the Tanaka and the Snyder“hydrophobic subtraction”tests;current theories and the effect of overload for ionised solutes;the use of high pH to improve peak shape;whether temperature is a useful parameter in improving peak shape;andfinally whether other sep-aration mechanisms such as HILIC can provide a viable alternative to RP–LC for the analysis of bases.2.Choice of column2.1.Column testing proceduresThe selection of an appropriate RP column for the analysis of bases can be a daunting task,as now many hundreds are com-mercially available,with a considerable number recommended especially by their manufacturers for the analysis of basic solutes. Nevertheless,several databases are now available where a large number of different columns have been subjected to the same test procedure by the same group of workers on the same or similar instruments,allowing a useful and objective comparison of perfor-860 D.V.McCalley /J.Chromatogr.A 1217(2010)858–880Fig.1.Structures present on a typical RP monomeric-bonded silica (C8)endcapped with trimethylsilyl groups.After U.D.Neue,“Silica Gel and its derivatization for Liquid Chromatography”,in “Encyclopedia of Analytical Chemistry”,R.A.Meyers,Ed.,John Wiley &Sons,Ltd.,Chichester (2000)11450–11472.mance to be made.A question arises as to the validity of databases constructed by evaluation of only a few or even a single column of a given type,as to whether the results obtained may be truly repre-sentative of the performance of this brand,due to column to column and batch to batch variations.However,a careful study [14,15]has suggested that columns from major manufacturers actually show a rather high degree of reproducibility,probably resulting from the use of stringent quality control procedures.Indeed,the industry is likely to be self-regulating to a degree,as dissatisfied customers would switch to the use of more reproducible brands.Tight reten-tion specifications exist in the HPLC user environment,especially in the pharmaceutical industry,and changes in the column can jeop-ardise product release.However,it is possible that a manufacturer could be forced to change the sourcing of a production raw mate-rial,which might occur for example,if the column manufacturer does not make their own silica.Thus,under some circumstances,a recently purchased column may not behave in the same way as one tested several years beforehand.Nevertheless,we believe that such situations are rare,and in most cases,manufacturers strive to main-tain the reproducibility of their products over a long period of time,as many customers have established methods on a given brand of phase.It appears more common to introduce a new name or name variant of an existing phase to mark definitively such changes or improvements to the production process.Taking this factor,and the reasonable reproducibility of commercial columns into account,it seems that the results of tests on a particular brand of column would generally reflect the performance of that brand throughout the product lifetime.Both of the column evaluation methods described in detail below incorporate strongly basic compounds as test probes.In each test,their retention is monitored at low and intermediate pH val-ues.Columns which give relatively low retention of basic probes are also likely to give higher efficiency for basic solutes,as shown by correlation studies for at least one of the procedures (see below).2.2.The Tanaka test and the Snyder hydrophobic subtraction parison of results with direct peak shape measurementsWhile many different column testing methods have been devel-oped,two have become prominent and have the distinct advantage that databases of results for many hundreds,rather than just a few columns,are available.The Tanaka method [16]and the hydropho-bic subtraction procedure developed by Snyder et al.[17]both incorporate tests which allow a user to select phases that are likely to be suitable for the separation of basic compounds.We will consider here the Tanaka method as adapted and applied by Euerby and Petersson [18]to the evaluation of over 200commercial columns that can be compared on a freely available program from Advanced Chemistry Development [19].These databases appear to be updated periodically;for instance,the ACD database contains evaluations of recently introduced sub-2m phases.An alternative adaptation of the Tanaka procedure and its application to a large number of different stationary phases has also been made [20],and data are again freely available [21].A fourth testing scheme is that published by the US Pharmacopeia.This protocol is an adaptation of the work of Sander and Wise [22].For activity towards bases,this method uses the tailing factor of amitriptyline (the same probe as used in the Snyder–Dolan procedure).At the time of writing,the database contained fewer columns than the two major proce-dures (∼100)and will not be considered further here.However,data for both this procedure and the Snyder–Dolan (S–D)method are available on the USP website [23].In the Tanaka–Euerby (T–E)procedure,columns are tested by measurement of k for pentylbenzene as a measure of sur-face area and surface coverage;hydrophobic selectivity from the ratio of k (pentylbenzene)/k (butylbenzene);shape selectivity from k (triphenylene)/k (o-terphenyl);hydrogen bonding capac-ity from k (caffeine)/k (phenol)in unbuffered methanol–water;total ion-exchange capacity from k (benzylamine)/k (phenol in methanol–phosphate buffer pH 7.6;and acidic ion-exchange capacity from k (benzylamine)/k (phenol)in methanol–phosphate buffer pH 2.7.The latter three tests are of particular interest for the analysis of basic solutes.The program [19]allows the comparison of the similarities and differences between various columns,and per-mits the separate weighting of the various factors—for example,columns can be ranked according solely to their total ion capacity at pH 7.6if so desired.The S–D model recognises that hydrophobic retention is the dominant process in RP chromatography,and in the absence of other retention mechanisms,plots of log k for one column versus another should be a straight line.However,these other mechanisms give rise to scatter in the plots.Clearly,ion-exchange and hydrogen bonding are important contributors to the retention of basic solutes.The general equation for retention in theD.V.McCalley/J.Chromatogr.A1217(2010)858–880861Table1Evaluation of some selected RP columns by two different procedures.For details on the procedure,see text.Column name k pentylbz k(pentbz)/k(butbz)k(triphen)/k(terph)k(caff)/k(phen)k(bzm)/k(phen)2.7k(bzm)/k(phen)7.6Tanaka–Euerby procedureChromolith 4.22 1.24 1.310.480.120.63Discovery Amide 1.65 1.35 1.810.490.190.44Discovery C18 3.32 1.48 1.510.390.100.28Inertsil ODS-37.74 1.45 1.290.480.010.29Resolve C18 2.40 1.46 1.59 1.29 1.23 4.06Spherisorb ODS-2 3.00 1.51 1.560.590.230.76Symmetry C18 6.51 1.46 1.490.410.010.68Symmetry Shield RP18 4.66 1.41 2.220.270.040.20Xterra MS C18 3.52 1.42 1.260.420.100.35Xterra RP18 2.38 1.29 1.830.330.070.20H S A B C(2.8)C(7.0)Snyder procedureChromolith 1.0030.0290.008−0.0140.1030.187 Discovery Amide0.7200.013−0.6250.218−0.092−0.025 Discovery C180.9840.027−0.1280.0040.1760.153 Inertsil ODS-30.9900.022−0.146−0.023−0.474−0.334 Resolve C180.968−0.1270.335−0.046 1.921 2.144 Spherisorb ODS-20.962−0.0760.070.0340.908 1.263 Symmetry C18 1.0520.0630.018−0.021−0.3020.123 Symmetry Shield RP180.8500.027−0.4110.093−0.7280.136 Xterra MS C180.9840.012−0.143−0.0150.1330.051 Xterra RP180.757−0.043−0.4830.097−0.170−0.173model is:log˛=log k/log k(ethylbenzene)=Á Hhydrophobic − S∗steric resistance(to bulky interactions)+ˇ Acolumn H-bond acidity(non-ionised silanols)+˛ BH-bond basicity(from sorbed water)+Ä Cion interaction(ionised silanols)(3)Ethylbenzene is used as a non-polar reference solute.Greek letters represent empirical,eluent-and temperature-dependent proper-ties of the solute,which are relative to the values for ethylbenzene, for which all solute parameters are identically zero.The selection of the optimum probes for evaluation of each retention mode has been made from detailed studies.Bold capitals represent eluent-and temperature-independent properties of the column;these val-ues are relative to a hypothetical average Type B C18column.Any column which behaves identically to this hypothetical reference column will have H=1and all other values S*,A,B,C=0.The dataset of columns evaluated by this procedure is even larger than that for the T–E procedure and presently extends to at least400columns.In some versions of the program,different weightings can be assigned to each evaluation parameter,as in the Euerby procedure.Results for some RP columns selected from each database are shown in Table1.The T–E data show clearly that the older Type A bonded phases(Resolve C18and Spherisorb ODS-2)give higher retention of benzylamine relative to phenol at pH7.6(alpha values 4.06and0.76,respectively)compared with newer Type B phases based on highly pure silica(Discovery C18and Inertsil ODS-3, alpha values0.28and0.29,respectively).Similarly with the S–D method,values of C(7.0)for Resolve C18and Spherisorb ODS-2 are high(2.144and1.263,respectively)compared with Discov-ery C18and Inertsil ODS-3(0.153and−0.334,respectively.Values of alpha(benzylamine/phenol)at pH2.7and values of C(2.8)are also higher for the Type A compared with the Type B phases using both procedures,indicating general agreement between them. Snyder and co-workers[24]have correlated a published dataset of“silanol activity”for a number of RP columns(measured by the average plate number for amitriptyline and pyridine with methanol-phosphate buffer pH6.0)with values of C at pH6.0,inter-polated from C(2.8)and C(7.0).Columns with a highvalue of C(6.0) correlated with columns of high silanol activity,and those with low values of C(6.0)with low silanol activity.In a later study[6]95%of Type B columns(designated either on the basis of manufacturer claims,or on the date a column wasfirst sold)were shown to have C(2.8)≤0.25,while only11%of Type A columns satisfied this crite-rion.Tailing of basic solutes(as measured by the asymmetry factor A s)was minimal for columns with C(2.8)<0.25(i.e.Type B columns) and tended to increase for larger values of C(2.8).From Table1,the Type A phases Resolve C18and Spherisob-ODS-2,now identified as such due to values of C(2.8)≥0.25,also give the highest values of hydrogen bonding acidity(parameter A,0.335and0.07,respec-tively,determined from the retention of amide probe compounds). Similarly,these phases also gave the highest relative retention of caffeine/phenol in the Tanaka procedure(1.29and0.59,respec-tively).The data can also be used to compare the effect of other features,e.g.the performance of embedded polar group phases (EPG)and the equivalent conventional C18phase,manufactured on the same silica.EPG phases include columns with embedded amide groups within the hydrocarbon chain:or carbamate groups:EPG phases have been proposed to give better peak shapes for the analysis of bases[24,27].The incorporation of an EPG in XTerra RP18reduces somewhat the Tanaka alpha(benzylamine/phenol) 7.6parameter to0.20,compared with0.35for the XTerra MS C18 column.Similarly,the S–D C(7.0)parameter is reduced to−0.173 for the EPG compared with0.051for the conventional phase.It is862 D.V.McCalley /J.Chromatogr.A 1217(2010)858–880possible that the reduced retention of benzylamine and other bases may be caused by a layer of water that is adsorbed close to the surface of EPG phases,providing some deactivating effect for the silanol groups [25,26].Other authors have compared conventional and EPG phases bonded on the same type of silica,on the basis of peak shape measurements.It was found that on average,peak shapes were indeed improved on the latter phases [27].Neverthe-less,it appears that the EPG technology gives more improvement in performance with phases bonded on older impure silicas,rather than the modern Type B silicas [27].This result seems to be reflected in the somewhat inconclusive data from Table 1concerning the rel-ative retention of bases on conventional and EPG phases.Thus the Discovery EPG phase (amide)has a slightly larger value of the T–E alpha (benzylamine/phenol)7.6parameter (0.44)compared with the regular C18phase (0.28).In contrast,the S–D C (7.0)parameter is smaller on Discovery Amide (−0.025)compared with Discovery C18(0.153).Similarly,while the T–E procedure indicates a con-siderable lower value of alpha (benzylamine/phenol)at pH 7.6for Symmetry Shield (0.2)compared with Symmetry C18(0.68),the S–D C (7.0)parameter for the EPG phases is slightly greater (0.136)compared with the regular phase (0.123).Euerby and Petersson pointed out that the extra retentiveness of phenols on EPG phases might invalidate the results of tests for silanophilic activity which involve the use of such solutes.They therefore suggested substitut-ing benzyl alcohol for phenol in the Tanaka test.Benzyl alcohol has retention properties similar to those of phenol but does not show excess retention on EPG phases [28].These particular comparisons point to some possible differences in the compatibility of column evaluations from either method.The Hoogmartens group looked more generally at the compati-bility of results from the S–D method and their own adaptation of the Tanaka procedure [29],finding a rather poor overall correlation between the two approaches.In a previous paper,this group had demonstrated a good correlation between their own method and the Euerby results.This latter finding is perhaps not surprising,as both are based on the Tanaka method.The problem of compatibility of the S–D and Tanaka methods may well be in the different mobile phase conditions and different probe solutes used in these tests.The S–D procedure uses the retention of the strong bases amitriptyline and nortriptyline in acetonitrile–phosphate buffer to calculate the cation-exchange term C (2.8)and derives the value of C (7.0)from the C (2.8)results by multiplying by the ratio of the retention fac-tors of the quaternary amine berberine at pH 7.0and 2.8;the T–E benzylamine tests use methanol as the organic modifier.Indeed the use of these different modifiers may explain the somewhat differ-ent evaluations of the EPG phases by either method.Even using the same mobile phase conditions,McCalley and Brereton [27,30–32]showed that peak shape data was not consistent between different basic probes.Thus,for example there was little correlation between A s for codeine and nortriptyline when using methanol–phosphate buffers at pH 3.0,whereas either of these solutes has been used as a single test compound to evaluate the relative silanol activity of different phases.One phase (Waters Symmetry Shield)gave,of 9highly inert RP columns,the highest N and lowest A s for nico-tine using acetonitrile–phosphate buffer at pH 7.0but the lowest efficiency for analysis of pyridine.Fig.2shows a principal compo-nents analysis (PCA)loadings plot for analysis of nine basic solutes on eight different RP columns using a mixture of methanol with a pH 3.0buffer.Lines can be drawn from the centre of the plot to each data point.Parameters that are opposed (i.e.appear at 180◦)measure equivalent but opposite trends.Thus N and A s values are opposed,with efficiency increasing as asymmetry decreases,as expected.Parameters that are at 90◦,like the asymmetry factors of pyridine and quinine,measure unrelated trends,and thus may be evaluating relatively different aspects of the detrimental inter-action of bases with the column surface.Conversely,the asymmetry parameters of nortriptyline and diphenhydramine have a smaller angle between them,and may be measuring more related proper-ties.It might therefore not be necessary to include both substances in a test mix for these particular mobile phase conditions.For over-all evaluation of column properties exploring different aspects of detrimental interactions,a test mix could include five compounds:codeine,quinine,amphetamine,nortriptyline and pyridine.The ranking of columns at pH 7using methanol was different from that at pH 7using acetonitrile;note that these correspond to the differ-ent modifiers of the T–E and S–D evaluation schemes,respectively.Snyder and co-workers [6]also observed that the tailing of basic (cationic)solutes on a given column appeared to be solute specific,finding that values of A s for the bases amitriptyline,nortriptyline,the quaternary compound berberine,and 4-n -hexylaniline corre-lated extremely poorly (r 2=0.01–0.19).The use of multiple basic test solutes and different mobile phase modifiers at different pH values would be a considerable task for the construction of these column evaluation databases.However,inclusion of a range of test compounds would undoubtedly improve the performance of these databases.It seems certain that these differences in test solutes and conditions contribute to the lack of correlation between the S–D and T–Etests.Fig.2.PCA loadings plots based on retention factor (k ),column efficiency (N ),Dorsey–Foley column efficiency (N df )and asymmetry factor (A s ).Data for eight different Type B reversed-phase columns and nine different probe compounds with methanol–phosphate buffer pH 3.0as mobile phase.See [30].。

摘要密级:学校代码:10075分类号:学号:********理学硕士学位论文TMPTA整体柱的制备及应用研究学位申请人:白晓梅指导教师:刘海燕副教授学位级别:理学硕士学科专业:药物分析学授予单位:河北大学答辩日期:二○一四年六月Classified Index: CODE: 10075 U.D.C.: NO: 20111327A Dissertation for the Degree of M. ScienceStudy on the Preparation andApplication of TMPTA MonolithicColumnsCandidate : Bai XiaomeiSupervisor : Asso. Prof. Liu HaiyanAcademic Degree Applied for : Master of ScienceSpecialty : Pharmaceutical AnalysisUniversity: Hebei UniversityDate of Oral Examination : June, 2014摘要近几年来,整体柱作为一种新型的高效液相色谱(HPLC)分离介质得到了快速发展,并且广泛应用于样品分析。
整体柱和传统的填充柱相比具有一定的优势,比如制备方法简单,传质速率快,渗透性高和易于表面修饰等。
本文首先以三羟甲基丙烷三丙烯酸酯(TMPTA)作为反应单体,偶氮二异丁腈(AIBN)为引发剂,乙二醇二甲基丙烯酸酯(EDMA)为交联剂,采用自由基原位聚合方法制备了有机聚合物整体柱。
考察了聚合条件的改变对孔结构的影响。
通过红外光谱、扫描电镜等方法对该整体柱的结构进行了表征,并且对其机械稳定性和渗透性进行了研究。
最后通过对一系列有机小分子的分离考察了目标整体柱的色谱保留行为。
其次,本文还制备了聚(三羟甲基丙烷三丙烯酸酯-N-异丙基丙烯酰胺-乙二醇二甲基丙烯酸酯)[poly(TMPTA-co-NIPAAm-co-EDMA)] 整体柱。
Jingwu KangDorothee WistubaVolker SchurigInstitute of Organic Chemistry, University of Tübingen,Tübingen,Germany A silica monolithic column prepared by the sol-gel process for enantiomeric separation by capillary electrochromatographyA method for the preparation of a silica monolithic capillary electrochromatography (CEC)column for the separation of enantiomers has been developed.The porous silica monolith was fabricated inside a fused-silica capillary column by using the sol-gel pro-cess.After gelation for24h,hydrothermal treatment at1007C for24h was performed to prevent the sol-gel matrix from cracking.The prepared monolith was then coated with Chirasil-b-Dex which represents a chiral polymer prepared by grafting permethyl-b-cyclodextrin to polymethylsiloxane with an octamethylene spacer.Immobilization of Chirasil-b-Dex was performed by heat treatment at1207C for48h to give a nonextract-able coating.The column performance was evaluated by using racemic hexobarbital as a model compound.The efficiency of9.26104theoretical plates/m for the first eluted enantiomer of hexobarbital was obtained at an optimal flow rate of the mobile phase.The effect of mobile phase composition on enantiomeric separation of hexo-barbital was also investigated.The column proved to be stable for more than one hun-dreds of runs during a two-months period.The enantiomers of several neutral and negatively charged chiral compounds were baseline separated on this column. Keywords:Capillary electrochromatography/Enantiomeric separation/Monolithic column/Sol-gel process EL48401IntroductionRecently,separation of enantiomers by capillary electro-chromatography(CEC)has received considerable atten-tion.Depending on the types of column design,three main approaches have been used for CEC enantiomeric separation:open tubular columns,packed columns and monolithic columns[1,2].Among these three approaches, the use of monolithic columns appears to be the most promising one because of the fritless column technology. The stationary phase of the monolithic column consists of a single piece that is chemically bonded on the inner wall of the capillary and therefore no supporting frit is required. Additionally,a monolithic column has an advantage of a high mass transfer and a low pressure drop.To date,several papers dealing with the preparation of monolithic CEC columns for enantiomeric separation have been published:(i)organic polymer-based mono-lithic columns[3–13];(ii)silica-based monolithic columns prepared by the sol-gel process[14–16]or by sintering the silica particles at high temperature[17].The organic polymer based method is characterized by simplicity, since the columns can be fabricated in one step by copolymerization of monomer and chiral selector.Silica-based monolithic columns have the advantage that the established silica modification methods used for prepar-ing HPLC chiral stationary phases can be directly trans-ferred to CEC.So far,only few papers concerning the preparation of monolithic column for CEC enantiomeric separation by the sol-gel technique have been published [14–16].Zare et al.[14]prepared a particle-loaded mono-lithic column for the enantiomeric separation of amino acids.Chen and Hobo[15]prepared a monolithic column by chemically modifying the silica sol-gel matrix with a ligand-exchange-type chiral stationary phase.Wang et al.[16]reported a method for the preparation of cyclo-dextrin based open tubular columns by the sol-gel tech-nique.Here,we describe a new method for the preparation of a cyclodextrin-based CEC monolithic column using the sol-gel process.The hydrothermal treatment was per-formed to prevent the monolith from cracking during the drying process.The prepared monolith was then modified with the chiral stationary phase Chirasil-b-Dex(Fig.1). The enantioselective column performance was evaluated and several neutral and negatively charged chiral com-pounds were successfully separated into enantiomers.Correspondence:Prof.Volker Schurig,Institute of OrganicChemistry,University of Tübingen,Auf der Morgenstelle18,D-72076Tübingen,GermanyE-mail:volker.schurig@uni-tuebingen.deFax:149-7071-295-538Abbreviations:SEM,scanning electron micrograph;TMOS,tet-ramethoxysilane1116Electrophoresis2002,23,1116–1120ªWILEY-VCH Verlag GmbH,69451Weinheim,20020173-0835/02/07-08–04–1116$17.50+.50/0C E a n d C E CFigure2.SEM of a sol-gel silica monolithic bed coated with Chirasil-b-Dex.Magnification,16506.different kind of stationary phases including chiral station-ary phases.However,the cracking of the wet gel during the drying process represents a drawback of this column technique[19,20].In order to alleviate this problem,a supercritical fluid drying technique was used in the litera-ture to prepare a cracking-free sol-gel-entrapped CEC column[20].In our hands,we found that the hydrothermal treatment was a very effective strategy to minimize the monolithic bed from extensive shrinking and to prevent it from cracking.This is probable due to the fact that hydro-thermal treatment promotes the further cross-linking of the residual silanol groups of the silica framework[21]. Figure2shows a scanning electronic micrograph(SEM) of a monolith prepared in our experiment.As can be seen,the morphology of the sol-gel matrix is reminiscent to a porous matrix formed by aggregated spherical silica particles.Chirasil-b-Dex was statically coated on the sur-face of silica matrix.Heat treatment at1207C was per-formed to result in a nonextractable coating of the immo-bilized chiral stationary phase.3.2Column performanceAs expected,the EOF velocity of the silica monolithic col-umn increased linearly with increasing the field strength in the range from0.1to0.6kV per cm.After modifying the silica monolith with the chiral stationary phase,the coat-ing of Chirasil-b-Dex shielded some of the silanol groups on the surface of silica,therefore a reduced EOF was obtained.However,the reduced EOF remained strong enough to drive the mobile phase through the capillary at an acceptable velocity when neutral analytes were investigated.For negatively charged analytes,a reversedFigure 4.Separation of enantiomers on the Chirasil-Dex monolithic column.Conditions:fused silica capillary column,25cm (effective length )650m m ID;UV de-tection was performed at 210nm.For mephobarbital,hexobarbital and benzoin:mobile phase,MES-Tris buf-fer (pH 6)/methanol (90/10v/v);applied field strength,0.4kV/cm;samples injection at 3kV for 4s;carprofen:mobile phase,MES-Tris buffer (pH 6)/methanol (60/40v/v);applied field strength,–0.4kV/cm;sample injection at –3kV for 4s.seen that the resolution (R s )is increased with a decrease of the flow rate,however,the analysis time is also in-creased.Therefore,a field strength of 0.4kV/cm (corre-sponding to a flow rate of 0.37mm/s)was selected for the further investigations.Several enantiomers including neutral and negatively charged chiral compounds were separated on this monolithic column.The chromato-grams for the baseline-separated enantiomers are shown in Fig.4.3.3Effect of mobile phase compositionon the separation of enantiomersThe effect of the methanol concentration in the mobile phase on the enantioselectivity was also investigated.As can be seen in Fig.5,the resolution factor R s decreased with raising the methanol concentration.Concurrently,an increase of the methanol concentration decreased the EOF velocity and prolonged the analysis time.Therefore,a low methanol concentration was selected.The effect of the buffer concentration on the separation of hexobar-bital enantiomers was also investigated at an MES con-centration range in the mobile phase from 20to 100m M .It was observed that the retention factor k increased slightly,however,the efficiency and resolution decreasedTable1.Reproducibility of elution time and resolution(R s) of hexobarbital enantiomersRun-to-run(n=5)Day-to-day(n=4)t1(min)t2(min)R s t1(min)t2(min)R s16.417.5 1.9216.1017.10 2.0216.317.3 2.0215.8416.96 1.9616.217.2 2.0713.7214.57 1.9916.117.1 2.0514.8015.77 2.1816.117.1 2.06RSD(%)0.880.9 2.327.207.35 4.81 Conditions:fused-silica capillary column,25cm(effective length)650m m ID;mobile phase,MES-Tris buffer(pH6)/ methanol(90/10v/v);applied field strength,0.4kV/cm; sample injection at3kV for4s.t1and t2are the elution times corresponding to the first and second eluted peaks of hexobarbital enantiomers.lithic bed as well as the Chirasil-b-Dex coating were proved to be stable.The reproducibility was evaluated via run-to-run(5runs)and day-to-day(4days).The results are summarized in Table1.A good run-to-run reproduci-bility was obtained on the same column.A relatively high deviation for day-to-day reproducibility could be due to the influence of the fluctuations of the ambient tempera-ture,since the used instrument was not thermostated.It should be noted that the day-to-day reproducibility was measured at random during two weeks.The column-to-column reproducibility was also evaluated,however,a rather poor result was obtained.Obviously,the reproduci-bility of the pore structure of the sol-gel matrix is poor between column-to-column,therefore,the thickness of the chiral polymer coating is difficult to control although the columns were coated with the same coating solution and thermally treated under the identical conditions.In conclusion,a method for the preparation of a porous sol-gel monolithic column used for the separation of enantiomers by CEC was developed.Hydrothermal treat-ment at1007C for24h was performed to prevent the silica monolith from cracking.After coating of the porous monolithic bed with Chirasil-b-Dex,the residual EOF remains strong enough to perform the separation at a reasonable time.The enantiomers of several racemic compounds were baseline-separated.The enantioselec-tive columns proved to be stable and showed a good run-to-run reproducibility.However,column-to-column reproducibility is rather poor.Further work towards the improvement of column-to-column reproducibility and enhancement of EOF is forthcoming.The authors thank Ms.Elke Nadler for SEM measure-ments and the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie for financial support. J.K.thanks the“Graduiertenkolleg Chemie in Inter-phasen”for a fellowship.Received August16,20014References[1]Wistuba,D.,Schurig,V.,Electrophoresis2000,21,4136–4158.[2]Lämmerhofer,M.,Sˇvec,F.,Fréchet,J.M.J.,Lindner,W.,Trends Anal.Chem.2000,19,676–698.[3]Peters,E.C.,Lewandowski,K.,Petro,M.,Sˇvec,F.,Fréchet,J.M.J.,mun.1998,35,83–86.[4]Lämmerhofer,M.,Peters,E.C.,Yu,C.,Sˇvec,F.,Fréchet,J.M.J.,Lindner,W.,Anal.Chem.2000,72,4617–4622. [5]Lämmerhofer,E.C.,Sˇvec,F.,Fréchet,J.M.J.,Lindner,W.,Anal.Chem.2000,72,4623–4628.[6]Hjertén,S.,Végvári,A.,Srichaiya,T.,Zhang,H.-X.,Ericson,C.,Eaker,D.,J.Capil.Electrophor.1998,5,13–26.[7]Végvári,Á.,Foldesi, A.,Hetenyi, C.,Kocnegarova,O.,Schmid,M.,Kudirkaite,V.,Hjertén,S.,Electrophoresis 2000,21,3116–3125.[8]Schmid,M.,Grobuscheck,N.,Tuscher,C.,Gübitz,G.,Vég-vári,Á.,Machtejevas,E.,Marusˇka,A.,Hjertén,S.,Electro-phoresis2000,21,3141–3144.[9]Koide,T.,Ueno,K.,J.Chromatogr.A2000,893,177–187.[10]Koide,T.,Ueno,K.,Anal.Sci.2000,16,1065–1070.[11]Koide,T.,Ueno,K.,Anal.Sci.1998,14,1021–1023.[12]Koide,T.,Ueno,K.,J.Chromatogr.A2001,909,305–315.[13]Schweitz,L.,Andersson,L.I.,Nilsson,S.,J.Chromatogr.A1997,792,401–409.[14]Kato,M.,Dulay,M.T.,Bennett,B.,Chen,J.-R.,Zare,R.N.,Electrophoresis2000,21,3145–3151.[15]Chen,Z.,Hobo,T.,Anal.Chem.2001,73,3348–3359.[16]Wang,Y.,Zeng,Z.,Guan,N.,Cheng,J.,Electrophoresis2001,22,2167–2172.[17]Wistuba,D.,Schurig V.,Electrophoresis2000,21,3152–3159.[18]Schurig,V.,Schmalzing,D.,Mühleck,U.,Jung,M.,Schlei-mer,M.,Mussche,D.,Duvekot,C.,Buyten,J.C.,J.High Resol.Chromatogr.1990,13,713–717.[19]Ishizuka,N.,Minakuchi,H.,Nakanishi,K.,Soga,N.,Naga-yama,H.,Hosoya,K.,Tanaka,N.,Anal.Chem.2000,72, 1275–1280.[20]Tang,Q.,Xin,B.,Lee,M.L.,J.Chromatogr.A1999,837,35–50.[21]Feng,P.Y.,Bu,X.H.,Pine,D.J.,Langmuir2000,16,5304–5310.1120J.Kang et al.Electrophoresis2002,23,1116–1120。