治疗疼痛的药物——阿片类镇痛药(3)
- 格式:doc
- 大小:387.00 KB
- 文档页数:32




阿片受体拮抗剂阿片受体拮抗剂本身对阿片受体并无激动效应,但对四受体有很强的亲和力,对K受体、8受体和。
受体也有一定的亲和力,可移除与这些受体结合的阿片类镇痛药物,从而产生拮抗效应。
目前的研究表明,阿片受体不仅存在于中枢神经系统,包括脑和脊髓,而且广泛存在于外周神经等部位。
一般的阿片受体拮抗剂全身应用对中枢和外周阿片受体均有作用,在拮抗阿片药物外周作用的同时,也减弱了中枢镇痛作用,主要包括纳洛酮、纳曲酮和纳美芬;而新型的外周阿片受体拮抗剂仅与外周阿片受体结合,与中枢阿片受体几乎不结合,可以拮抗阿片药物的外周作用,但不减弱阿片药物的中枢镇痛效应,主要包括甲基纳曲酮和Alvimopan。
一、纳洛酮(naloxone)纳洛酮又名N-烯丙去甲羟基吗啡酮(N-allyl-noroxymorphone)。
结构式为:H0分子式:C19H21NO4分子量:327.21纳洛酮拮抗阿片类药物的强度是烯丙吗啡的30倍,对中枢和外周阿片受体均有效,不仅可拮抗吗啡等纯阿片受体激动药,而且可拮抗喷他佐辛等阿片受体激动-拮抗药,但对丁丙诺啡的拮抗作用稍弱。
纳洛酮的亲脂性很强,约为吗啡的30倍,易于透过血-脑脊液屏障。
静脉注射后脑内药物浓度可达血浆浓度的4.6倍,而吗啡脑内浓度仅为血浆浓度的1/10。
纳洛酮的分布容积为1.81 L/kg,与血浆蛋白结合率为46%,主要在肝内与葡萄糖醛酸结合后随尿排出,清除率14〜30 ml/(kg-min)。
消除半衰期30〜78分钟。
由于在脑内的浓度下降迅速,故药效维持时间短。
静脉注射后2〜3分钟即可产生最大效应,作用持续时间约45分钟;肌内注射后10分钟产生最大效应,作用持续时间约2.5〜3小时。
纳洛酮主要应用于主要用于:①拮抗阿片药物急性中毒的呼吸抑制;②全麻的手术结束后,用以拮抗阿片药物的残余作用;③娩出的新生儿因受其母体中阿片药物影响而致呼吸抑制,可用纳洛酮拮抗;④纳洛酮可激发阿片药物成瘾者的戒断症状,具有诊断价值。

第二节阿片类镇痛药一、作用机制阿片类镇痛药又称麻醉性镇痛药( narcotic analgesics ),就是一类能消除或减轻疼痛并改变对疼痛情绪反应的药物。
除少数作用弱的药物以外,此类药物若使用不当多具有成瘾性,但用于医疗目的并不会带来太大问题。
研究显示慢性疼痛患者长期采用阿片类药物治疗时,成瘾的发生率极低。
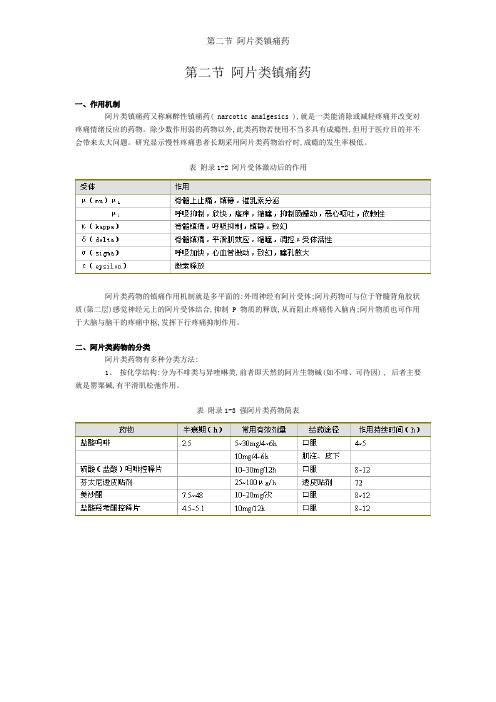
表附录1-2 阿片受体激动后的作用阿片类药物的镇痛作用机制就是多平面的:外周神经有阿片受体;阿片药物可与位于脊髓背角胶状质(第二层)感觉神经元上的阿片受体结合,抑制 P 物质的释放,从而阻止疼痛传入脑内;阿片物质也可作用于大脑与脑干的疼痛中枢,发挥下行疼痛抑制作用。
二、阿片类药物的分类阿片类药物有多种分类方法:1、按化学结构:分为不啡类与异喹啉类,前者即天然的阿片生物碱(如不啡、可待因) , 后者主要就是罂粟碱,有平滑肌松弛作用。
表附录1-3 强阿片类药物简表表附录1-4 弱阿片类药物简表2、按来源该类药物可分为天然阿片类、半合成衍生物 ( 如双氢可待因,二乙酰不啡 ) 与合成的阿片类镇痛药。
合成药物又分为四类:①苯丙不啡烷类 (phenylpiperidine derivatives) ,如哌替啶、芬太尼等;②不啡喃类 (morphinenans) ,如左不喃;③苯异不啡烷类 (bengmorphans) ,如喷她佐辛;④二苯甲烷类 (diphenylmethanes) ,如美散酮。
3、按受体类型可分为μ、κ、δ受体,该三种受体的分子结构已被确定,并被成功克隆。
从功能上还可能存在ε与δ受体,并可能进一步分为μ 1 、μ 2 、κ 1 、κ 2 、κ 3 与δ 1 、δ 2 等亚型。
表 3-2 为受体激动后的药理作用。
4、按药理作用分,阿片类镇痛药又可分为激动药 ( 不啡、芬太尼、哌替啶等 ) ,激动一拮抗药( 喷她佐辛、纳布啡等 ) ,部分激动药(丁丙诺啡)与拮抗药 (纳洛酮等) 。
激动—拮抗药又称部分激动药,主要激动κ受体,对δ受体也有一定激动作用,而对μ受体则有不同程度的拮抗作用。

阿片类镇痛药第一节:概述:疼痛是一种因组织损伤或潜在的组织损伤而产生的痛苦感觉,常伴有不愉快的情绪甚或心血管和呼吸方面的变化。
——它既是机体的一种保护性机制,提醒机体避开或处理伤害,也是临床许多疾」病的常见症状.剧烈疼痛不仅给患者带来痛苦和紧张不安等情绪反应,还可引起机体生理功能紊乱, 甚至诱发休克。
控制疼痛是临床药物治疗的主要目的之躯体痛(Somaticpai n):快痛、慢痛,对机械性、化学性、炎症性、温度性刺激均敏感。
内脏痛(Visceral pai n):对牵张、炎症刺激敏感。
神经痛(Neuropat hi c pai n):多由神经损伤或兴奋性增高引起,呈发作性或持续性,一般镇痛药无效。
躯体疼痛分两类:快痛(剧痛):(Fast pain, acute pain, sharp pai n)尖锐而定位清楚的刺痛,刺激时立即发生,撤除刺激立即消失。
由A S -类纤维传导。
慢痛(钝痛)(chro ni c pa in, sl ow pa in, blunt pain)定位不明确的烧灼痛,发生较慢,持续时间较长。
疼痛除了使患者感受痛苦外,常伴有情绪、心血管和呼吸等方面的变化。
剧痛常引起失眠或其它生理机能的紊乱,甚至引起休克。
由无髓鞘的G类纤维传导。
阿片那与疼痛产生有关的物质:绝大多数情况下,伤害性神经末稍的有效刺激即为化学物质, 包括: 神经递质类(5-HT 、组胺、ACh )、激肽类(缓激肽、赖氨酰缓激肽)、 代谢产物(ATP 、ADP 、H+、K+)、 前列腺素类(PGE2)、 辣椒素(Capsai ci n )。
与疼痛传导有关的神经递质和调质:许多物质参与了痛觉信号的传递和调控过程,包括: 神经肽类:P 物质(SP )、神经激肽(NKA 、NKB ) 经典递质类:Gu )、GA3A 、5 — HT 、NA 腺嘌呤.阿片肽类(opi oi d pept i des )亮氨酸脑啡肽、甲硫氨酸脑啡肽、内啡肽、强啡肽、 内吗啡肽等。

![阿片类镇痛药[内容充实]](https://img.taocdn.com/s1/m/08ec3c4e87c24028915fc3db.png)





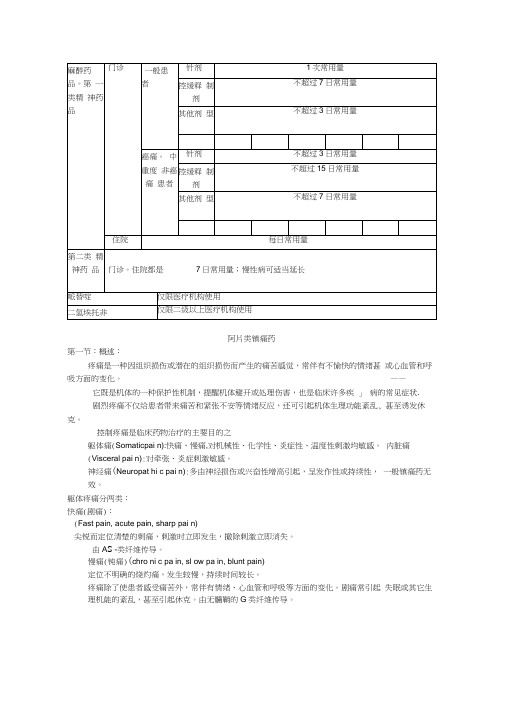
癌症患者止痛的三阶梯治疗癌症疼痛是一个普遍性的问题,有效的止痛治疗是世界卫生组织癌症综合规划四项重臾之一。
1982年世界卫生组织为实现“到2000年让癌症患者不痛,并提高其生活质量”的目标,在全球推行癌痛治疗计划。
卫生部于1991年下达了关于我国开展“癌症患者三级止痛阶梯治疗方案”工作的通知,以及镇痛药临床应用的五项基本原则。
所谓癌痛治疗的三阶梯方法就是在对癌痛的性质和原因做出正确的评估后,根据患者的疼痛程度和原因适当地选择不同作用强度的镇痛药。
第一阶梯的药物为非留体抗炎药,代表药物为阿司匹林, 其他药物有对乙酰氨基酚、布洛芬、双氯芬酸、高鸟甲素、蔡普生以及口引躲美辛栓(肛内)等(见骨骼肌和风湿免疫疾病用药)。
这类药物主要用于轻、中度疼痛的患者,也可作为第二、第三阶梯的辅助用药。
第二阶梯的药物为弱阿片类镇痛药,代表药物为可待因, 其他药物有双氢可待因、氨酚待因、氢可酮、羟考酮、布桂嗪、曲马多等。
这类药物主要用于中度疼痛的患者或第一阶梯用药后仍有疼痛的患者。
第三阶梯的药物为强效阿片类镇痛药,代表药物为吗啡, 其他药物有氢吗啡酮、羟吗啡酮、左啡诺、二氢埃托啡、美沙酮、芬太尼等。
这类药物主要用于重度疼痛的患者或应用了第二阶梯的药物后疼痛仍不能缓解的患者。
1、癌症疼痛药物治疗的主要原则(1) 口服给药:首选口服给药,口服给药经济、方便,尤其对强效阿片类镇痛药不易产生依赖性,这样便于患者长期用药。
若不适合口服给药或达不到止痛效果,可采用透皮贴剂、肛门给药和输液泵连续皮下用药。
(2)按时给药:按照药物的有效作用时间有规律地按时给药,而不是按需给药,这样才能使患者维持恒定的有效血药浓度,以达到使癌症患者不痛的目的。
(3)按阶梯给药。
(4)个体化用药:所用药物剂量是应以能使患者达到有效镇痛为准,不应以各种镇痛药物推荐的常规剂量为标准, 也不受药典中规定的“极量”的限制。
一方面因为药物作用存在个体差异;另外,在长期使用阿片类药物的过程中,每个人的耐受情况不同,调整剂量也会有区别。
第二节吗啡吗啡为纯天然阿片类生物碱。
1806年德国化学家F.W.A.Serturner从鸦片中提出纯品吗啡;1847年Knorr确定其分子式;上世纪20年代初J.M.Gulland和R.Robinson提出吗啡的化学结构;1952年M.Gates和G.Tschudi人工合成了吗啡,才正式确定了其化学结构。
虽历经一百余年,吗啡仍然是目前使用最为广泛的阿片类药物之一,因其止痛效果确切、价格低廉而被世界卫生组织(WHO)推荐为阿片类镇痛药物的标准用药,通常也作为其他阿片类药物临床评估的参考。
化学结构:分子式:C17H19NO3分子量:285.37一、药动学吗啡口服易吸收,由于肝脏和消化道的首过效应(first-pass effect),其生物利用度为30%~40%,控缓释制剂与即释制剂生物利用度相近,直肠给药生物利用度变异比较大。
皮下、肌肉和静脉注射吗啡无首过效应,生物利用度接近100%。
吗啡的pKa为7.9,进入血管内的吗啡约有1/3与血浆蛋白结合,其余在生理pH下呈游离状态,并迅速分布于人体大多数组织中,包括实质性脏器、空腔脏器甚至体液、皮肤、毛发等,其表观分布容积(apparent volume of distribution,Vd)约为1.0~4.7 L/kg。
只有游离状态的吗啡可通过血-脑脊液屏障和胎盘组织。
吗啡的辛醇/水(octanol/water)分配系数在pH7.4时为1.42,呈低脂溶性,亲水性强,虽然吗啡在机体内分布广泛,但其组织穿透力弱,因而只有极少部分吗啡(静脉注射后不到1%)能够透过血-脑脊液屏障发挥镇痛作用。
吗啡血浆清除率(plasma clearance)为0.9~1.2 L/(kg·h),主要在肝脏代谢。
5%左右去甲基化生成去甲吗啡(normorphine),60%~80%葡萄糖醛酸化,其分子结构上3位和6位的羟基分别被葡糖醛酸替代,生成吗啡-3-葡萄糖醛酸(morphine-3-glucuronide,M3G)和吗啡-6-葡萄糖醛酸(morphine-6-glucuronide,M6G)。
M3G是吗啡的主要代谢产物,无药理学活性,其本身并不具药理拮抗作用,但可影响吗啡的代谢,使其不能作用于阿片受体,从而显著减弱吗啡及M6G 的镇痛作用。
M6G约占代谢产物的10%,外周给予M6G,其镇痛效能是吗啡的3~5倍,而鞘内给药其镇痛效能是吗啡的10~60倍,作用时间较吗啡稍长。
这与M6G透过血-脑脊液屏障能力大约只有吗啡的1/57有关。
M6G易通过胎盘。
吗啡透过血-脑脊液屏障发挥中枢镇痛作用,一部分游离吗啡再从脑脊液缓慢释放到体循环;另有少部分可能经脑组织直接代谢生成M6G。
脑脊液中极少量M6G 即可发挥镇痛效应,包括呼吸抑制。
因此,硬膜外和鞘内注射吗啡可出现中枢延迟性呼吸抑制。
吗啡的代谢产物和一些游离吗啡,主要经肾脏排出,此外还可经乳汁和胆汁等途径排泄,其终末半衰期一般在1.5~4 小时。
在肾功能不全的患者,可以导致M6G的蓄积,使吗啡作用时间延长。
在肝功能障碍、即便早期肝昏迷的患者,葡萄糖醛酸化作用很少受到损害,吗啡的代谢基本不受影响。
早产儿的肝脏已经具有代谢吗啡的能力,但吗啡葡萄苷酸化的能力受到妊娠时间以及产后体重和年龄的影响。
荟萃分析结果表明,吗啡在儿童的分布容积为2.8±2.6 L/kg,似乎于年龄无关,而消除半衰期和血浆清楚率与年龄相关。
早产儿、0~57天的新生儿和11~15岁的婴幼儿及儿童的血浆清除半衰期分别为9.0±3.4 h、6.5±2.8 h和2.0±1.8 h,血浆清除率分别为2.2±0.7 ml/(kg•min)、23.6±8.5 ml/(kg•min)和8.1±3.2 ml/(kg•min)。
空腹时口服即释吗啡30 分钟起效,饱胃状态则起效延迟,达峰效应需1~2 小时,作用维持4~5 小时。
长效吗啡制剂使得口服吗啡越来越方便,药动学更为稳定。
国内可使用的长效吗啡口服制剂包括盐酸吗啡控释片(美菲康)和硫酸吗啡控释片(美施康定),其起效时间较即释吗啡稍慢但不受食物影响,需较长时间才能达到峰效应(平均3.7 小时),血浆峰浓度较即释吗啡低,作用可以有效持续8~12 小时。
硫酸吗啡缓释胶囊(Kadian)使用聚合酶涂层,作用可持续24 小时。
直肠给药同样生物利用度变异较大,其起效时间、达峰效应时间和作用持续时间与口服吗啡相近,美施康定直肠给药与口服等效。
单次静脉注射吗啡数分钟即可起效,30 分钟达峰效应,作用持续2~3 小时。
皮下和肌内注射吗啡除起效稍慢外,止痛作用及副反应与静脉途径相当,但使用更方便,其峰效应时间为45~90 分钟,作用持续3~4 小时。
吗啡静脉PCA 时,血浆最低有效止痛浓度约为20~40 ng/ml。
硬膜外腔注射吗啡3 mg,硬膜外血管丛迅速吸收,注射后5~10 分钟即达血浆峰浓度,平均33~40ng/ml,血浆终末消除半衰期90±34.3 min。
约不到1/10吗啡缓慢透过硬脊膜进入脑脊液,其吸收半衰期平均为22 min,需60~90分钟达脑脊液峰浓度,手术后止痛的脑脊液最小有效浓度平均150 ng/ml。
硬膜外注入2~6 mg的吗啡后,脑脊液吗啡峰浓度相当血浆浓度的50~250倍,随即呈双相消除,半衰期分别为1.5 小时(快速消除相)和6 小时(缓慢消除相),说明脑脊液里吗啡亦难透过血-脑脊液屏障返回体循环,其有效作用时间可达20 小时。
新的硫酸吗啡延缓释放酯质体注射剂(morphine sulfate extended-release liposome injection,商品名DepoDur)10 mg注入硬膜外腔后缓慢释放进入体循环和透过硬脊膜进入脑脊液,血浆峰浓度平均仅为20 ng/ml,消除半衰期长达16 小时左右,可以有效止痛达36~48 小时。
鞘内注入0.3 mg吗啡,血浆峰浓度时间5~10 分钟,平均血药浓度4.5±1.1 ng/ml,脑脊液浓度6410±1290 ng/ml。
吗啡注入脑脊液经15~30 分钟的快速分布相浓度开始下降,消除半衰期为89.8±16.1 min。
注射后6 小时脑脊液内吗啡的平均浓度仍达332±137 ng/ml。
吗啡鞘内初始分布容积约为22±8 ml。
吗啡脑脊液药动学具有明显个体差异,个体之间持续作用时间长短不一。
二、药效动力学1.中枢神经系统吗啡的中枢神经作用同其他阿片类药物,包括镇痛、镇静、镇咳以及呼吸抑制、瞳孔缩小、恶性呕吐、尿潴留等。
(1)镇痛镇静:吗啡选择性激活脊髓胶质区、丘脑内侧、脑室及导水管周围灰质的阿片受体,产生强大的镇痛作用。
吗啡可有效缓解种疼痛,对持续性慢性钝痛的效力大于间断性锐痛。
吗啡还有明显的镇静作用。
吗啡激动边缘系统和蓝斑核的阿片受体,改善疼痛所引起的焦虑、紧张、恐惧等情绪反应,伴有欣快感并出现嗜睡、精神朦胧、神志障碍等,安静时易诱导入睡,但易唤醒。
大剂量吗啡(15~20 mg)镇痛镇静作用更明显,且无封顶效应。
(2)镇咳:直接抑制咳嗽中枢,使咳嗽反射减轻或消失。
(3)抑制呼吸:治疗量吗啡可降低呼吸中枢对血液CO2张力的敏感性和抑制脑桥呼吸调整中枢,使呼吸频率减慢,潮气量降低。
随着吗啡剂量增大,呼吸抑制作用增强。
急性中毒时呼吸频率可减至3~4次/分,最后呼吸停止,这是吗啡急性中毒致死的主要原因。
(4)缩瞳:吗啡主要通过兴奋支配瞳孔的副交感神经引起瞳孔缩小。
阿片类药物过量时瞳孔缩小,针尖样瞳孔为其中毒特征,但不具有特异性(例如桥脑出血或缺血性损伤后亦可出现同样体征)。
吗啡过量呼吸抑制导致严重的低氧血症时可以出现显著的瞳孔散大。
阿片类药物的缩瞳作用可以耐受,因而在长期使用阿片类药物的患者,其注射吗啡缩瞳作用减弱。
(5)其他:吗啡直接兴奋延脑呕吐中心化学感受器导致恶心、呕吐。
此外,吗啡通过抑制下丘脑释放促性腺释放激素、促肾上腺皮质激素释放因子及抗利尿激素的释放2.平滑肌(1)胃肠道:吗啡一方面兴奋胃肠平滑肌,提高其肌张力,减缓推进性蠕动, 使内容物通过延缓和水分吸收增加;另一方面吗啡可以提高回盲瓣及肛门括约肌张力,使肠内容物通过受阻。
同时吗啡还通过中枢抑制作用,减弱便意和排便反射,引起便秘,用于止泻。
(2)胆道:吗啡收缩胆道奥迪括约肌,使胆道排空受阻,导致上腹部不适甚至引起胆绞痛(阿托品可部分缓解)。
(3)支气管:大剂量吗啡时可导致支气管平滑肌时收缩,诱发和加重哮喘。
(4)膀胱:吗啡提高膀胱括约肌的张力,导致排尿困难,尿潴留。
(5)子宫:吗啡降低子宫张力,对抗催产素对子宫的收缩作用,延长产程,产妇禁用。
3.心血管系统(1)扩张血管及降低外周血管阻力:这与吗啡抑制血管运动中枢、促进组胺释放有关。
有时可引起体位性低血压。
(2)间接扩张脑血管:吗啡引起的呼吸抑制,CO2潴留可使使脑血管扩张,导致颅内压升高。
因此颅脑损伤,颅内高压患者禁用。
4.其他抑制免疫系统和HIV诱导的免疫反应。
一次给予吗啡后后早期出现短暂的IL-1、IL-2、TNF-α 、TNF-β 和IL-10增加,随后出现持久下降,直到给药后48 小时才恢复。
吗啡还抑制NK细胞活性、抑制刀豆素A刺激T细胞增殖及抑制巨噬细胞吞噬功能。
三、临床应用1.镇痛主要用于各种原因导致的中重度急性疼痛和慢性癌痛。
可有效缓解或消除严重创伤、烧伤、手术、心肌梗死等引起的剧痛;目前不主张作为缓解胆道平滑肌痉挛绞痛(需加用解痉药如阿托品、长托宁等)一线药。
吗啡长期使用仅限于缓解癌痛。
(1)口服即释吗啡主要用于某些急性疼痛的短期治疗和癌性爆发痛的控制,吗啡控缓释制剂仅限于于中至重度癌痛的治疗,原则上从小剂量开始,最好在24~72 小时内滴定至较理想止痛用药剂量。
以美施康定为例,初始剂量30 mg,12或24 小时时评价患者疼痛强度(V AS,10分),如V AS≥7分,剂量增加50%~100%;V AS 5~6分,剂量增加25%~50%;V AS≤4分则增加25%的剂量。
须注意:滴定剂量应同时调整按时给药和必要时给药的用量;备用阿片类即释片作为必要时用药;疼痛V AS评分<4分及不良反应严重时减量;如果用药剂量突然较明显变化,应重新评估疼痛及病情并考虑是否产生耐药。
美施康定可以直肠给药,用于不能口服的患者,其剂量滴定同口服。
(2)不推荐长期间断使用静脉、皮下和肌内注射吗啡用于缓解癌痛。
有文献报道,持续吗啡静脉和皮下PCA给药用于缓解终末期癌痛取得良好疗效。
(3)植入式电子微量注射泵鞘内给药是目前效力最高,全身副反应最小的阿片类药物的给药途径,经典药物仍使用吗啡。