四个基团相互不等同,则是一手性中心,如
果连有一对相同基团时,该碳原子则是前
手性中心.一般来说前手性中心与手性中心相连,那么这一对相同的基团肯定是化学不等价.如果不与手性中心相连则用对
称面原则来判断,若存在对称面,两个基团
则是对映异位的.反之则是非对映异位.O
H
H3C
CH(CH3)2CH34.同一碳相同二基团a).固定在环上CH2两个氢不是化学等价
如2.分子内存在着快速运动R4R5R1R2R3
R6常见的分子内存在有链的旋转,环的翻转.由于分子内的快速运动,一些不能通过对称操作而交换的基团有可能为化学等价,但也不是两个
相同的基团就一定成为化学等价基团. RCH2-CXYZY
X
R Ha
Z
Y
X
Hb R
Z
X
Ha HbY
R
Hb
Z
Ha从分子旋转的角度,分子总是处于1,2,3 三种构象之一,当温度升高,链的旋转速度加大,三种构象的分子逐步
接近,当无论如何,Ha与Hb也不能是化学等价的.如果把R=H,三个氢是完全等价的.所以甲基的三个氢总是在同一位置.3.前手性(prochirality)在有机化合物中,如果与某碳原子相连的
=5.25+0.8+0.11+0=6.16(实际测定6.10Hb=5.25+Z同CH2Br)+Z顺CO2R)+Z反H)=5.25+0.70+1.18+0=7.13(实际测定7.10)苯环质子化学位移的计算取代苯环的氢化学位移可按照下式计算:δ=7.26+Σ Zi 7.26是未取代的苯环的δ值, Zi是取代参
的核相互偶合但不与体系外任何一个核
偶合.在体系内部不要求一个核和它以外所有的核都偶合.例如CH3COOC2H5分别存在A3和A3X2两个自旋体系.2.谱图分类的原则1).分子中化学位移相同而且对外偶合常数也相同(磁等价),用一个大写英文字母表示,如A1,A2,A3….,下标为核的数目.
2).分子中化学位移不同的核用不同的大写英文字母表示.如果核之间的化学位移之差Δν与J数值相当,用AB,ABC,ABCD….表示,如果Δν比J大许多(Δν/J>6),用AX,AMX,AMPX…表示.3).化学等价但磁不等价的核用AA’,BB’表示表2.3 一些分子自旋体系和波谱类型F
2-4 1,4-
1,9 2.3自旋偶合体系(spin system)2.3.1化学等价(chemical equivalence)化学等价是立体化学中的一个重要观念.
如果分子中两个相同原子(或两相同基团)
处于相同化学环境时,它们是化学等价.化学不等价的两个基团,在化学反应中,可以反映出不同的反应速度,在光谱,波谱的测量中,可能有不同的测量结果,因而可用谱学方法来研究化学等价性.1。考察分子各原子核相对静止状态可用对称操作分析两个基团能否相互交
分子中氢的种类和数目就可以非常容易地推断出有机物的分子结构。2.1 化学位移化学位移是核磁共振最重要参数之一.前
面我们已经讨论了影响化学位移的因素.这里不再讨论.根据上述各种影响氢核化学位移的因素和多年核磁共振测定有机物结构的经验同样总结出了不同有机基团氢核的化学位移δ值。根据δ值可
以进行相应有机基团的推断常见的一些有机基团的氢核的化学位移总结于表2.1中。常见基团化学位移H
用经验公式计算.这些经验公式是根据取代基对化学位移地影响具有加和性(additivity)的原理由大量实验数据归纳总结出来的.某些情况下估算具有较高准确度,具有实用价值,而在某些场合下,虽
然误差较大,但依然有参考价值.化学位移计算主要目的是:1).对谱线进行归属;2).为测定分子结构提供理论依据.亚甲基与次甲基的δ计算对于亚甲基可以用Shoolery公式加以计
CH4Cl2CHCHO
CH3CH2OH
Ph-CH=CH23.核磁共振的谱图的分类核磁谱图可分为一级谱图和二级谱图.一级谱图表现满足两个条件:Δν/J>6同一核组的核必须是磁等价.一级谱图有以下特点:峰的数目可用n +1 规律描述.峰的强度可用二项式展开系数表示.从谱图中可以直接读出δ,J.峰的中心位置为δ,相邻两峰之间的距离为J.1.5
换来判断两个基团(核)是否化学等价.可分为三种情况.两个取代基完全相同,Ha,Hb可以用二次
对称轴C2和对称平面相互交换.具有相同的化学位移,它们是化学等价的. CHa
X
XHbO
H
H3C
CH(CH3)2CH3两个取代基不同,但可以用对称平面,或
者二次旋转对称轴联系起来,具有相同的化学位移,它们是化学等价的.反之则是化学不等价.例
苯6.08.5
醛RCHO 9.010.0
羧酸RCOOH 10.512.0酚4.012.0对于大部分有机化合物来说氢谱的化学位移值在0-13 ppm. 大致可分以下几个区0-0.8 ppm 很少见典型化合物; 环丙烷硅烷以及金属有机化合物。0.8-1.5 ppm烷烃区域. 氢直接与脂肪碳相连没有强电
负性取代基。化学位移地次序
CH>CH2>CH3.。如果有更多的取代基化学位移移向低场。1.5-2.5 ppm 羰基区域质子相邻羰基C=O, C=C or 苯环。3.0-4.5 ppm 醚区域. (同样醇酯有CH-O group.) 质子直接邻氧如果有更多的电负性取代基化学位移移向低场。5.0-7.0 ppm 双键区域. 氢直接与C=C 双键相连. 7.0-8.0 ppm 芳环质子区域. 磁各向异性作用导致芳环质子处于去屏蔽区。同样现象发生在醛由于羰基地磁各向异性醛质子化学位移在
9-10 ppm
-OH 可以出现在任何位置谱线的性质由多重
因此影响H的交换pH.浓度温度溶剂等。
一般芳环酚羟基更趋于低场。
大多数的-NHR, -NH2和醇一样可被交换在
2-3 ppm 区域显示宽峰。
-CO2H 可交换,象醇(>11 ppm) 化学位移的计算某些基团或化合物的质子化学位移可以
R顺
R反
H
同RδC=C-H=5.25 + Z同+Z顺+Z反Z同,Z顺,Z反分别代表相应取代基的取代参数.参阅宁永成P40`41C C
Cl
HCl
Fδ=5.25 +1.08 +0.18 -1.02 =5.49 (5.56)Hb
CH2Br
H3COOC
HaHa=5.25+Z同CO2R)+Z顺CH2Br)+Z反H)
的.例如O
Ha
Hb
H
HΔδ=0.39ppmb).单键不能快速旋转,同碳上两个基团是不等价的.由于C-N单键具有双键性质,不能自由旋转,氮上两个甲基是化学不等价的.
N
CH3CH3
O
H
3CC).与手性碳相连的CH2的两个氢是不等价的.2.3.2磁等价(magnetic equivalence)两个核磁等价必须满足下列两个条件:它们是化学等价的它们对任意另外一个核的偶合常数相同
炔 C≡CH 2.03.0
苯基取代2.23.0
醚基取代ROCH33.34.0溴取代CH3Br 2.54.0氯取代CH3Cl 3.04.0
羟基取代CH3OH 4.04.3氟取代CH3F 4.04.5
酰氧基取代RCOOCH33.74.1胺RNH21.05.0醇ROH 1.05.5烯CCH 4.65.9
J (Hz)
类型
J ( Hz)
12-15 2-9
0 6,5- 7,5
5,5-
7,0
aa 5-8
ae 2-4
ee 2-4 0,5- 3 7-1213-18 4-10
0,5-
2,5
0
9- 13 2- 3
1- 3 2- 4
o 6-
-COR
-COOH0.8
0.9
1.32.01.9
1.4
1.7
1.5
2.3
2.7
2.9
1.0
1.0
3.01.01.2
1.2
0.8
0.7
1.2
C C对于次甲基的δ值依然可以用Shoolery
经验公式计算,但常数项改为1.5. δ=1.50 +Σσ烯烃的化学位移计算C C
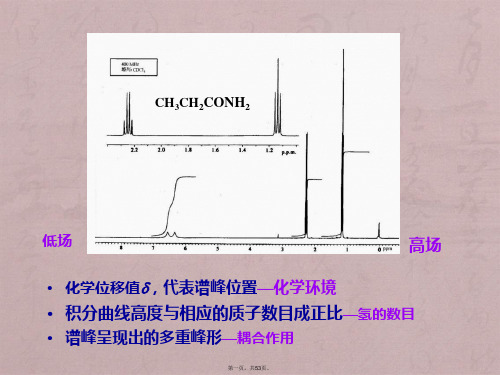
图中横坐标为化学位移,图上有三个峰则表明该有机物分子中的氢有三种类型
峰面积的积分比为952表明该化合物的三种不同氢的数目分别是9、5和2化学位移δ 7.2处的峰表示苯环上5个相同的氢δ2.5处的峰表示亚甲基上的2个相同氢而δ0.9处的峰则表示三个甲基上的9个相同的氢。这样能够判断出有机物
Δν/J
6.5
5.5
4.5
2.5
算δ=1.25 +Σσ (2-1)式中σ为取代基的经验屏蔽常数.表中给出其数值.表2.2 Shoolery 公式中的经验屏蔽常数(σ)取代基
σ
RC=C-Ph
Cl
Br
I
OH
-OR-OPh-OCOR
-OCOPh
NH2
NR2
NO2SR-CHO
9
m 1-
3
p 0-
1
1-2 1,6-
2,0
1-3 0,6-
1,0
1-4 1,3-
1,8
2-3 3,2-典型nJH,H-偶合常数1-2 2,0-
2,6
1-3 1,5-2,2 1-4 1,8-
2,3
2-3 2,8-
HCH3HOH氢核类型
果连有一对相同基团时,该碳原子则是前
手性中心.一般来说前手性中心与手性中心相连,那么这一对相同的基团肯定是化学不等价.如果不与手性中心相连则用对
称面原则来判断,若存在对称面,两个基团
则是对映异位的.反之则是非对映异位.O
H
H3C
CH(CH3)2CH34.同一碳相同二基团a).固定在环上CH2两个氢不是化学等价
如2.分子内存在着快速运动R4R5R1R2R3
R6常见的分子内存在有链的旋转,环的翻转.由于分子内的快速运动,一些不能通过对称操作而交换的基团有可能为化学等价,但也不是两个
相同的基团就一定成为化学等价基团. RCH2-CXYZY
X
R Ha
Z
Y
X
Hb R
Z
X
Ha HbY
R
Hb
Z
Ha从分子旋转的角度,分子总是处于1,2,3 三种构象之一,当温度升高,链的旋转速度加大,三种构象的分子逐步
接近,当无论如何,Ha与Hb也不能是化学等价的.如果把R=H,三个氢是完全等价的.所以甲基的三个氢总是在同一位置.3.前手性(prochirality)在有机化合物中,如果与某碳原子相连的
=5.25+0.8+0.11+0=6.16(实际测定6.10Hb=5.25+Z同CH2Br)+Z顺CO2R)+Z反H)=5.25+0.70+1.18+0=7.13(实际测定7.10)苯环质子化学位移的计算取代苯环的氢化学位移可按照下式计算:δ=7.26+Σ Zi 7.26是未取代的苯环的δ值, Zi是取代参
的核相互偶合但不与体系外任何一个核
偶合.在体系内部不要求一个核和它以外所有的核都偶合.例如CH3COOC2H5分别存在A3和A3X2两个自旋体系.2.谱图分类的原则1).分子中化学位移相同而且对外偶合常数也相同(磁等价),用一个大写英文字母表示,如A1,A2,A3….,下标为核的数目.
2).分子中化学位移不同的核用不同的大写英文字母表示.如果核之间的化学位移之差Δν与J数值相当,用AB,ABC,ABCD….表示,如果Δν比J大许多(Δν/J>6),用AX,AMX,AMPX…表示.3).化学等价但磁不等价的核用AA’,BB’表示表2.3 一些分子自旋体系和波谱类型F
2-4 1,4-
1,9 2.3自旋偶合体系(spin system)2.3.1化学等价(chemical equivalence)化学等价是立体化学中的一个重要观念.
如果分子中两个相同原子(或两相同基团)
处于相同化学环境时,它们是化学等价.化学不等价的两个基团,在化学反应中,可以反映出不同的反应速度,在光谱,波谱的测量中,可能有不同的测量结果,因而可用谱学方法来研究化学等价性.1。考察分子各原子核相对静止状态可用对称操作分析两个基团能否相互交
分子中氢的种类和数目就可以非常容易地推断出有机物的分子结构。2.1 化学位移化学位移是核磁共振最重要参数之一.前
面我们已经讨论了影响化学位移的因素.这里不再讨论.根据上述各种影响氢核化学位移的因素和多年核磁共振测定有机物结构的经验同样总结出了不同有机基团氢核的化学位移δ值。根据δ值可
以进行相应有机基团的推断常见的一些有机基团的氢核的化学位移总结于表2.1中。常见基团化学位移H
用经验公式计算.这些经验公式是根据取代基对化学位移地影响具有加和性(additivity)的原理由大量实验数据归纳总结出来的.某些情况下估算具有较高准确度,具有实用价值,而在某些场合下,虽
然误差较大,但依然有参考价值.化学位移计算主要目的是:1).对谱线进行归属;2).为测定分子结构提供理论依据.亚甲基与次甲基的δ计算对于亚甲基可以用Shoolery公式加以计
CH4Cl2CHCHO
CH3CH2OH
Ph-CH=CH23.核磁共振的谱图的分类核磁谱图可分为一级谱图和二级谱图.一级谱图表现满足两个条件:Δν/J>6同一核组的核必须是磁等价.一级谱图有以下特点:峰的数目可用n +1 规律描述.峰的强度可用二项式展开系数表示.从谱图中可以直接读出δ,J.峰的中心位置为δ,相邻两峰之间的距离为J.1.5
换来判断两个基团(核)是否化学等价.可分为三种情况.两个取代基完全相同,Ha,Hb可以用二次
对称轴C2和对称平面相互交换.具有相同的化学位移,它们是化学等价的. CHa
X
XHbO
H
H3C
CH(CH3)2CH3两个取代基不同,但可以用对称平面,或
者二次旋转对称轴联系起来,具有相同的化学位移,它们是化学等价的.反之则是化学不等价.例
苯6.08.5
醛RCHO 9.010.0
羧酸RCOOH 10.512.0酚4.012.0对于大部分有机化合物来说氢谱的化学位移值在0-13 ppm. 大致可分以下几个区0-0.8 ppm 很少见典型化合物; 环丙烷硅烷以及金属有机化合物。0.8-1.5 ppm烷烃区域. 氢直接与脂肪碳相连没有强电
负性取代基。化学位移地次序
CH>CH2>CH3.。如果有更多的取代基化学位移移向低场。1.5-2.5 ppm 羰基区域质子相邻羰基C=O, C=C or 苯环。3.0-4.5 ppm 醚区域. (同样醇酯有CH-O group.) 质子直接邻氧如果有更多的电负性取代基化学位移移向低场。5.0-7.0 ppm 双键区域. 氢直接与C=C 双键相连. 7.0-8.0 ppm 芳环质子区域. 磁各向异性作用导致芳环质子处于去屏蔽区。同样现象发生在醛由于羰基地磁各向异性醛质子化学位移在
9-10 ppm
-OH 可以出现在任何位置谱线的性质由多重
因此影响H的交换pH.浓度温度溶剂等。
一般芳环酚羟基更趋于低场。
大多数的-NHR, -NH2和醇一样可被交换在
2-3 ppm 区域显示宽峰。
-CO2H 可交换,象醇(>11 ppm) 化学位移的计算某些基团或化合物的质子化学位移可以
R顺
R反
H
同RδC=C-H=5.25 + Z同+Z顺+Z反Z同,Z顺,Z反分别代表相应取代基的取代参数.参阅宁永成P40`41C C
Cl
HCl
Fδ=5.25 +1.08 +0.18 -1.02 =5.49 (5.56)Hb
CH2Br
H3COOC
HaHa=5.25+Z同CO2R)+Z顺CH2Br)+Z反H)
的.例如O
Ha
Hb
H
HΔδ=0.39ppmb).单键不能快速旋转,同碳上两个基团是不等价的.由于C-N单键具有双键性质,不能自由旋转,氮上两个甲基是化学不等价的.
N
CH3CH3
O
H
3CC).与手性碳相连的CH2的两个氢是不等价的.2.3.2磁等价(magnetic equivalence)两个核磁等价必须满足下列两个条件:它们是化学等价的它们对任意另外一个核的偶合常数相同
炔 C≡CH 2.03.0
苯基取代2.23.0
醚基取代ROCH33.34.0溴取代CH3Br 2.54.0氯取代CH3Cl 3.04.0
羟基取代CH3OH 4.04.3氟取代CH3F 4.04.5
酰氧基取代RCOOCH33.74.1胺RNH21.05.0醇ROH 1.05.5烯CCH 4.65.9
J (Hz)
类型
J ( Hz)
12-15 2-9
0 6,5- 7,5
5,5-
7,0
aa 5-8
ae 2-4
ee 2-4 0,5- 3 7-1213-18 4-10
0,5-
2,5
0
9- 13 2- 3
1- 3 2- 4
o 6-
-COR
-COOH0.8
0.9
1.32.01.9
1.4
1.7
1.5
2.3
2.7
2.9
1.0
1.0
3.01.01.2
1.2
0.8
0.7
1.2
C C对于次甲基的δ值依然可以用Shoolery
经验公式计算,但常数项改为1.5. δ=1.50 +Σσ烯烃的化学位移计算C C
图中横坐标为化学位移,图上有三个峰则表明该有机物分子中的氢有三种类型
峰面积的积分比为952表明该化合物的三种不同氢的数目分别是9、5和2化学位移δ 7.2处的峰表示苯环上5个相同的氢δ2.5处的峰表示亚甲基上的2个相同氢而δ0.9处的峰则表示三个甲基上的9个相同的氢。这样能够判断出有机物
Δν/J
6.5
5.5
4.5
2.5
算δ=1.25 +Σσ (2-1)式中σ为取代基的经验屏蔽常数.表中给出其数值.表2.2 Shoolery 公式中的经验屏蔽常数(σ)取代基
σ
RC=C-Ph
Cl
Br
I
OH
-OR-OPh-OCOR
-OCOPh
NH2
NR2
NO2SR-CHO
9
m 1-
3
p 0-
1
1-2 1,6-
2,0
1-3 0,6-
1,0
1-4 1,3-
1,8
2-3 3,2-典型nJH,H-偶合常数1-2 2,0-
2,6
1-3 1,5-2,2 1-4 1,8-
2,3
2-3 2,8-
HCH3HOH氢核类型










![核磁共振氢谱[1] (2) (1)](https://img.taocdn.com/s1/m/e52d2a23b4daa58da0114a1f.png)


