药物与血浆蛋白结合的程度
- 格式:ppt
- 大小:1.31 MB
- 文档页数:55

血浆蛋白结合药物的原理
血浆蛋白结合药物的原理是指药物与血浆中的蛋白分子结合形成药物-蛋白复合物。
主要是药物与血浆中的白蛋白结合,也可以与其他血浆蛋白如α1-酸性糖蛋白、β-球蛋白等结合。
血浆蛋白结合药物的原理由以下几个方面解释:
1. 提高药物的溶解度:药物结合血浆蛋白后,可以增加药物在血浆中的溶解度,提高药物的稳定性,避免药物在血浆中沉淀或析出。
2. 调控药物的分布:结合血浆蛋白后,药物的分布范围受到限制。
药物-蛋白复合物主要分布在血浆中,而不易进入组织或细胞内。
这可以延长药物在体内的半衰期,增加药物的持续时间,提高药物的疗效。
3. 调节药物的代谢和排泄:血浆蛋白结合药物经过代谢和排泄时,药物必须与蛋白分离才能被肝脏或肾脏清除。
这种结合可以延长药物在体内的循环时间,并减慢药物从体内被清除的速度。
总之,血浆蛋白结合药物的原理是通过与血浆蛋白结合形成药物-蛋白复合物,提高药物的溶解度,调节药物的分布范围和代谢排泄,从而影响药物的疗效和药物在体内的代谢动力学。

药物的血浆蛋白结合是指药物进入循环后首先与血浆蛋白结合,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,联合药物的药理活性暂时消失。
当分子膨胀时,结合物不能通过毛细血管壁,所以它们可以暂时“储存”在血液中。
药物与血浆蛋白结合的特异性较低,但其结合位点有限。
两种药物可能与同一种蛋白质竞争替代品。
例如,蛋白酶与双香豆素竞争血浆蛋白,这会增加血浆中的自由形式浓度,从而可能导致出血。
这种组合的特点如下可逆性结合后,药物活性暂时消失:结合物变大,不能通过毛细血管壁暂时储存在血液中,不经分布和消除。
可能发生竞争性取代:药物与血浆蛋白结合的特异性较低,但血浆蛋白结合位点有限。
两种药物可能与同一种蛋白质竞争产生替代品。
结合率如下药物的血浆蛋白结合能力受药物浓度、血浆蛋白质量和数量、解离常数的影响。
药理学书籍记载的药物血浆蛋白结合率是正常人在正常剂量范围内测得的值。
影响药物血浆蛋白结合率的因素和后果如下当两种药物同时使用时,会发生竞争性替代。
如果一种药物的结合率达到99%,就会被另一种药物取代,降低1%,理论上游离(药理活性)药物的浓度将增加100%,可能导致中毒。
然而,在游离药物不断增加的过程中,血浆中的药物浓度会不断增加。
药物也可能与内源性代谢物竞争并与血浆蛋白结合,如磺胺类药物交换胆红素和血浆蛋白结合,这可能导致新生儿核黄疸。
当血浆蛋白过低(如肝硬化)或恶化(如尿毒症)时,药物的血浆蛋白结合率降低,容易引起毒性反应。
由于血浆蛋白有限,当结合率高的药物在结合部位达到饱和时,如果药物剂量继续增加,血浆中的游离药物浓度将大大增加,引起毒性反应。


药物的血浆蛋白结合是指药物进入循环后首先与血浆蛋白结合,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,结合药物的药理活性暂时消失。
由于分子扩大,结合物不能通过毛细血管壁,因此它们可以暂时“储存”在血液中。
药物与血浆蛋白结合的特异性低,但是血浆蛋白的结合位点受到限制。
两种药物可能会与同一种蛋白质竞争替代品。
例如,宝泰松与双香豆素竞争血浆蛋白,这会增加血浆中的游离型浓度,这可能导致出血。
组合的特征如下可逆性结合后,药理活性暂时消失:结合物变大,无法通过毛细血管壁暂时储存在血液中,而没有分布和消除。
可能发生竞争性替代:药物与血浆蛋白结合的特异性低,但血浆蛋白的结合点有限。
两种药物可能会与同一蛋白质竞争而产生替代现象。
结合率如下药物的血浆蛋白结合能力受药物浓度,血浆蛋白的质量和数量以及解离常数的影响。
药理学书籍中记录的药物血浆蛋白结合率是在正常剂量范围内对正常人测得的值。
影响药物血浆蛋白结合率的因素及后果如下当两种药物一起使用时,就会发生竞争性替代。
如果一种药物的结合率达到99%,则当其被另一种药物替代并降低1%时,理论上游离型(具有药理活性)药物的浓度将增加100%,这可能导致中毒。
但是,在普通药物的更换过程中,游离药物将被迅速清除,血浆中游离药物的浓度难以持续增加。
药物也可能与内源性代谢物竞争并与血浆蛋白结合,例如磺酰胺交换胆红素和血浆蛋白结合,这可能导致新生儿发生核黄疸。
当血浆蛋白过少(如肝硬化)或变质(如尿毒症)时,药物血浆蛋白的结合率降低,容易引起毒性反应。
由于血浆蛋白有限,当具有高结合率的药物在结合位点达到饱和时,如果药物剂量继续增加,血浆中游离药物的浓度将大大增加并引起毒性反应。

实验二药物血浆蛋白结合率测定前言:药物血浆蛋白结合率是药物与血浆蛋白结合的量占药物总浓度的百分率,是药物代谢动力学的重要参数之一。
它影响药物在体内的分布、代谢与排泄,从而影响其作用强度和时间,并往往与药物的相互作用及作用机制等密切相关。
研究药物血浆蛋白结合率的方法包括常规的平衡透析法、超滤法、超速离心法、凝胶过滤法、分配平衡法、稳定同位素-GC-MS法、光谱技术等。
本实验主要通过采用平衡透析法对药物血浆蛋白结合率进行测定,对于新药研究开发和指导临床合理用药都具有重要意义。
关键词:血浆蛋白结合率平衡透析法磺胺嘧啶【实验原理】平衡透析法的基本原理是将蛋白置于一个隔室内,用半透膜将此隔室与另一隔室隔开。
蛋白等大分子不能通过此半透膜,但系统中游离配基可自由通过。
当达到平衡时半透膜两侧自由配基的浓度相等。
若系统中自由配基的总量已知,测定不含蛋白隔室中自由配基的浓度,即可推算与蛋白结合的配基量。
再用重氮化偶合比色测定法对磺胺药的含量进行测定。
其测定原理是具有游离氨基的磺胺药在酸性介质中重氮化后,可与N-(1萘基)乙二胺产生偶合反应生成紫红色偶氮染料,再与同样处理的磺胺药标准液比较,即可求得剩余的亚硝酸影响测定,用氨基磺酸胺分解除去。
【材料与试剂】1. 药物10%磺胺嘧啶钠注射液2. 试剂15%三氯醋酸溶液0.1%亚硝酸钠溶液0.5%氨基磺酸胺溶液0.1%二盐酸N-(1萘基)-乙二胺溶液磺胺嘧啶钠贮存标准液磺胺嘧啶钠应用标准液(3.75ug/ml)3.透析液pH7.4的0.02M 磷酸盐缓冲液,内含0.15M NaCl4. 鸡、兔各3只5. 管状半透膜周长5cm,长约12cm【试剂的配制】1. 0.1%二盐酸N-(1萘基)-乙二胺溶液0.5 g溶于95 %乙醇约400 mL中,再用乙醇稀释至500ml,棕色瓶于冰箱中保存2.贮存标准液取标准品0.125g溶于蒸馏水中,并稀释至1000mL。
如不溶于水,可先溶于0.1 mol/L氢氧化钠25mL中,再加4mol/L硫酸175 mL,用蒸馏水稀释至1000mL。



药物的血浆蛋白结合是指药物进入循环后首先与血浆蛋白结合,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,联合药物的药理活性暂时消失。
当分子膨胀时,结合物不能通过毛细血管壁,所以它们可以暂时“储存”在血液中。
药物与血浆蛋白结合的特异性较低,但血浆蛋白结合位点有限。
两种药物可能与同一种蛋白质竞争替代品。
例如,蛋白酶与双香豆素竞争血浆蛋白,这会增加血浆中的自由形式浓度,从而可能导致出血。
这种组合的特点如下可逆性结合后,药物活性暂时消失:结合物变大,不能通过毛细血管壁暂时储存在血液中,不经分布和消除。
可能发生竞争性取代:药物与血浆蛋白结合的特异性较低,但血浆蛋白的结合位点有限。
两种药物可能与同一种蛋白质竞争产生替代物。
结合率如下药物的血浆蛋白结合能力受药物浓度、血浆蛋白质量和数量以及解离常数的影响。
药理学书籍记载的药物血浆蛋白结合率是正常人在正常剂量范围内测得的值。
影响药物血浆蛋白结合率的因素和后果如下当两种药物同时使用时,会发生竞争性替代。
如果一种药物的结合率达到99%,被另一种药物取代并降低1%,理论上游离(药理活性)药物的浓度将增加100%,可能导致中毒。
但在更换常用药物的过程中,游离药物会迅速被清除,血浆中游离药物浓度难以持续升高。
药物也可能与内源性代谢物竞争并与血浆蛋白结合,如磺胺类药物交换胆红素和血浆蛋白结合,这可能导致新生儿核黄疸。
当血浆蛋白过低(如肝硬化)或恶化(如尿毒症)时,药物血浆蛋白结合率降低,容易引起毒性反应。
由于血浆蛋白有限,当结合率高的药物在结合部位达到饱和时,如果药物剂量继续增加,血浆中游离药物浓度将大大增加,引起毒性反应。

药物与血浆蛋白结合该药物的血浆蛋白结合情况如下药物进入循环后,首先成为与血浆蛋白结合的药物,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,偶联药物的药理活性暂时消失。
由于分子的膨胀,结合物不能通过毛细血管壁,所以它们可以暂时“储存”在血液中。
药物与血浆蛋白的结合特异性较低,但血浆蛋白的结合位点有限。
两种药物可能会与同一种蛋白质争夺替代品。
如保泰松与双香豆素争夺血浆蛋白,增加了后者的游离型浓度,可能导致出血。
结合特点:可逆结合后药理活性暂时消失;结合物变大,不能通过毛细血管壁暂时储存在血液中,不能分布和消除。
可能出现竞争性替代:药物与血浆蛋白结合的特异性较低,但血浆蛋白结合点有限。
两种药物可能与同一种蛋白质竞争,产生替代现象。
结合率如下药物浓度、血浆蛋白的质量和数量以及解离常数影响药物的血浆蛋白结合能力。
药理学书籍中记录的药物血浆蛋白结合率是正常人群在常用剂量范围内测量的值。
影响药物血浆蛋白结合率的因素及后果如下当两种药物同时使用时,出现竞争性替代。
如果一种药物的结合率达到99%,被另一种药物取代后降低1%,理论上游离型(具有药理活性)药物的浓度就会增加100%,可能导致中毒。
然而,在普通药物被替代的过程中,游离药物会迅速被淘汰,血浆中游离药物的浓度难以持续升高。
药物也可能与内源性代谢物与血浆蛋白结合,如磺胺类药交换胆红素和血浆蛋白结合,这可能会导致新生儿核黄疸。
当血浆蛋白过少(如肝硬化)或病情恶化(如尿毒症)时,药物血浆蛋白结合率降低,容易引起毒性反应。
由于血浆蛋白的限制,当结合率高的药物在结合位点达到饱和时,如果药物剂量继续增加,血浆中游离药物的浓度会大大增加,引起毒性反应。

司美格鲁肽蛋白结合率司美格鲁肽(Sitagliptin)是一种口服降糖药物,属于双肽酶-4(DPP-4)抑制剂。
它通过抑制DPP-4酶的活性,从而延长肠道激素胰高血糖素样肽-1(GLP-1)和胰岛素释放多肽(GIP)的作用时间。
这些肠道激素在胰岛细胞中起着重要的降低血糖作用,因此,司美格鲁肽主要通过促进胰岛细胞分泌胰岛素、抑制胰岛葡萄糖异生以及通过降低胃肠道葡萄糖吸收而达到降低血糖的效果。
蛋白结合率是药物在体内与血浆蛋白结合的百分比,也是一个重要的药理参数。
它描述了药物与蛋白质的亲和力和结合效果,对药物的分布、代谢和排泄等药物动力学过程具有重要影响。
在司美格鲁肽中,蛋白结合率是衡量其药理特性之一。
理解司美格鲁肽的蛋白结合率对于了解其药代动力学特性以及临床应用具有重要意义。
首先,让我们来了解一下蛋白结合的意义。
蛋白结合率的高低会直接影响药物在血浆中的自由浓度,从而影响其活性和作用方式。
当药物与血浆蛋白结合的越多,药物在血浆中的自由浓度越低,对靶组织的作用可能会相对减弱。
相反,当药物与血浆蛋白结合的越少,药物在血浆中的自由浓度越高,对靶组织的作用可能会相对增强。
对于司美格鲁肽而言,它主要与血浆蛋白里面的白蛋白结合。
白蛋白是一种广泛存在于体内的蛋白质,具有很高的结合能力。
根据研究,司美格鲁肽的蛋白结合率约为38%至40%左右。
这意味着大约有60%至62%的司美格鲁肽在血浆中是未与蛋白结合,以自由的形式存在。
该蛋白结合率对于司美格鲁肽的临床应用产生了一些影响。
首先,蛋白结合率较低的司美格鲁肽可以更容易地通过血脑屏障,与DPP-4酶发生作用。
这一效果有助于司美格鲁肽对中枢神经系统的作用,从而进一步调节血糖。
其次,蛋白结合率较低还可以增加司美格鲁肽的自由浓度,提高其在体内的活性和药效。
另外,需要注意的是,司美格鲁肽的蛋白结合率可能受到其他因素的影响。
例如,一些药物可能与司美格鲁肽竞争与蛋白质的结合位点,降低其蛋白结合率,从而影响其药效。

药物与血浆蛋白结合的药理学研究对于药物和血浆蛋白的结合,通常的认知是二者形成的复合体是不能够实现跨膜运转的,进而使机体摄入的药物在分布、代谢、排泄及与相应的受体在结合后产生的药理效果,会以一种游离的形式进行,而游离药物在血液中发生的浓度变化是对机体内药物处置、药效起到决定性影响的因素中的重要一种。
本文对药物与血浆蛋白结合的药理学基础和研究进展进行阐述,对临床的常规用药需要考虑的因素进行总结,从而能够明确药物在何种情况下需要监测游离的浓度。
1药物与血浆蛋白的结合机体在使用药物后,药物进入机体内循环,由于结构上的差异性,就会与血细胞、血浆蛋白互相结合,形成结合型药物,而没有发生结合的药物,被称为游离药物。
药物在不同的作用下进行结合,这些作用分别是共价键结合、离子键吸引、氢键结合、电荷转移、疏水性结合及范德华引力,而上述的药物结合方式是可逆的,如果结合后,药物分子发生变大的情况,就不易透膜,能够在血液中储存,然后经过血液运输,通过游离药物分布到机体的各个组织部位,进而起到治疗的作用。
在血液中,多数药物能够与血浆蛋白结合,能够与血细胞结合,或者进入血细胞,当进入血细胞后,分布在血液中,继而形成了一种动态的平衡。
但在上述的各种结合方式中,药物与血浆蛋白的结合会对药物的分布造成重要的影响,特别是药物结合成的蛋白组分中,白蛋白(HSA)和α1酸性糖蛋白(AGP)是2种最为重要的。
1.1结合方式为保证药物的安全性和临床用药的有效性,人们在研究中,将药物与血浆蛋白的结合规律进行了重点研究,研究发现,药物与血浆蛋白的结合方式,对患者体内的药物浓度起到预测的作用,根据质量守恒的作用原理,药物和血浆蛋白的可逆结合要保证以下的平衡:ka=k1/k-1=1/kd(1)式(1)中,k1是结合速率常数,解离速率常数是k-1,药物-蛋白复合物结合常数是ka,解离常数是kd。
该公式反应的是药物与蛋白在结合产生的亲和力的大小。
高蛋白与药物结合的ka值范围在105~107mmol/L,而低蛋白结合的ka值和中等结合强度ka的值范围均在102~104mmol/L。
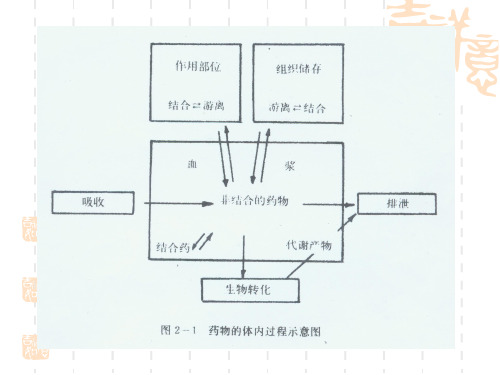

血浆蛋白结合率对药物作用的影响
血浆蛋白结合率:药物进入血液后与血浆蛋白结合的量占血液总药量的比例。
各种药物以一定的比率与血浆蛋白结合,在血浆中常同时存在结合型与游离型。
而只有游离型药物才具有药物活性。
药物与血浆蛋白结合成为结合型药物,暂时失去药理活性,并“储存”于血液中,起到药库的作用。
对于药物作用及其维持时间长短有重要意义。
结合分为可逆性、饱和性、非特异性、竞争性。
药物与血浆蛋白的结合影响药物在体内的分布、转运速度以及作用强度和消除速率。
一般蛋白结合率高的药物体内消除慢,作用维持时间长,药效平稳。
结合率低的药物体内消除快,同时作用时间短,药效有很大的波动。
药物内源性性化合物也可在血浆蛋白结合部位发生竞争性置换作用,两种以上的药物联用时,可相互竞争血浆蛋白的结合部位,结合力强的药物能从蛋白结合部位上取代结合力弱的药物,使后者游离型数量增加,导致药效和毒性反应亦增强。
其影响程度可因后者在体内的分布容积不同而异。
一般只有血浆蛋白结合率高,分布容积小,消除慢以及治疗指数低的药物在临床上的这种相互作用才有意义。


药物与血浆蛋白结合药物与血浆蛋白结合率的变化通过影响游离型药物浓度,改变药物分布、代谢、排泄以及作用靶点的结合,从而影响药理效应及毒副作用。
对1500种常用药的研究表明,43%化合物的血浆蛋白结合大于90%。
血浆蛋白结合在不同的治疗领域没有显著差异:中枢神经系统、炎症和肾-心血管。
唯一的例外可能是抗炎药物,其高蛋白结合者(>99%)占较高的比例(26%),且其中大多数为酸类。
令人惊奇的是,许多中枢神经系统药物具有高血浆蛋白结合的特点。
该研究最惊人的发现是,化学治疗药物(包括抗生素、抗病毒药物、抗真菌药物和抗肿瘤药物)中有较高比例(77%)的低结合药物。
在设计化学治疗药物方面,低血浆蛋白结合似乎更有优势。
血浆蛋白结合的基本原理血液经抗凝处理后的全部血液为全血;血浆是血液(不包括细胞)的液体部分。
在存在抗凝剂(如肝素)的条件下采集新鲜血液,然后离心去除血细胞而得到用于体外研究的血浆,血浆蛋白仍然保留于液体部分。
血清是去除了凝血因子(如纤维蛋内原)的血浆,它是在不含抗凝剂条件下采集的。
药动学研究通常采集血浆,从而保留了蛋白结合型及游离型药物,但除去了与细胞结合的药物。
许多药物与血浆蛋白、组织蛋白或体内的大分子物质如白蛋白、DNA 等反应,生成药物大分子复合物。
药物与蛋白类高分子结合后,分子体积变大,不能透过血管壁向组织转运,不能由肾小球滤过,不能透过胎盘屏障,不能经肝代谢。
进人血液中的药物,一部分在血液中呈游离形式存在,一部分与血浆蛋白通过离子键、氢键、疏水键及范德华力可逆性结合,形成药物血浆蛋白复合物,是药物在血浆中的一种贮存形式,能降低药物的分布与消除速度,维持药物疗效。
药物的蛋白结合不仅影响药物的体内分布,而且还影响药物的代谢和排泄。
药物在机体内最重要的结合就是蛋白结合,且主要是血浆蛋白结合。

药物与血浆蛋白结合
血液经抗凝处理后的全部血液为全血;
血浆是血液(不包括细胞)的液体部分。
在存在抗凝剂(如肝素)的条件下采集新鲜血液,然后离心去除血细胞而得到用于体外研究的血浆,血浆蛋白仍然保留于液体部分。
血清是去除了凝血因子(如纤维蛋内原)的血浆,它是在不含抗凝剂条件下采集的。
药动学研究通常采集血浆,从而保留了蛋白结合型及游离型药物,但除去了与细胞结合的药物。
许多药物与血浆蛋白、组织蛋白或体内的大分子物质如白蛋白、DNA 等反应,生成药物大分子复合物。
药物与蛋白类高分子结合后,分子体积变大,不能透过血管壁向组织转运,不能由肾小球滤过,不能透过胎盘屏障,不能经肝代谢。
进人血液中的药物,一部分在血液中呈游离形式存在,一部分与血浆蛋白通过离子键、氢键、疏水键及范德华力可逆性结合,形成药物血
浆蛋白复合物,是药物在血浆中的一种贮存形式,能降低药物的分布与消除速度,维持药物疗效。
药物的蛋白结合不仅影响药物的体内分布,而且还影响药物的代谢和排泄。
药物在机体内最重要的结合就是蛋白结合,且主要是血浆蛋白结合。
药物的血浆蛋白结合率是影响药物分布的重要因素。
血液中结合型的药物不易透过细胞膜,只有游离型的药物可向血管外扩散,转运到组织中。
如果药物的血浆蛋白结合率高,血浆中游离型药物浓度少,进入其他组织的浓度就低。
因此,药物分布主要取决于血液中的游离型药物的浓度,另外也与该药物和组织结合程度有很大关系。
蛋白结合对药物分布的影响见表4-5,可见血浆中游离型药物浓度越高,越容易向其他组织转运,药物的表观分布容积越大。

血浆是一种水溶液,包含92%的水,7%的蛋白质以及1%的其它物质(比如无机盐),总共占到人体血液的55%的组分。
药物与血浆蛋白的结合(Plasma Protein Binding, PPB)会降低血液循环系统中自由药物浓度,从而影响渗透到组织细胞到达药物靶点的治疗能力,或者影响肾脏的消毒能力。
因此,PPB的结合事件会影响药物在体内的活性代谢时间和毒副作用,患者的其他药物,食物和病理状况的共同给药可以显着改变药物的结合百分比,并可能导致严重后果。
药物分子可以通过多种方式与血浆蛋白结合,而且血浆蛋白结合作用是可逆的,主要取决于疏水作用和静电作用,比如vdW作用和氢键。
血浆蛋白结合药物与游离态药物的浓度达成动态平衡,这个可逆平衡过程会强烈影响各种药理学性质,比如分布体积,药物清除率,和消除,以及药理效应。
因为只有一部分游离药物可以跨过细胞膜,高蛋白结合能力的药物比低蛋白结合能力的药物有更长的半衰期。
结合到血浆蛋白上的药物越多,发挥治疗作用的游离药物的比例越小。
血浆蛋白对药物的结合能力是评估药动力学特点的一个非常重要的性质,评估游离药物在组织器官和血液中的比例是药物设计中的一个有价值的科学问题。
血浆蛋白主要包括白蛋白、球蛋白、凝血因子和调节蛋白,最重要的药物结合蛋白是白蛋白和α1-酸性糖蛋白,其次是脂蛋白。
其中,白蛋白的浓度最高(600μM),是血浆中的主要药物结合组分(人类血清白蛋白,HSA,human serum albumin);其次是α-酸性糖蛋白(AAG,α-acid glycoprotein,α-也叫做乳清类粘蛋白),12-30μM,和脂蛋白(lipoproteins,也叫做γ-球蛋白)。
关于药物结合HSA和AAG的研究工作是最多的。
白蛋白是人类血浆蛋白中的主体组分(600 μM),含量占到总血浆蛋白的60%。
白蛋白(HSA),是人血浆中含量最丰富的一种蛋白质,能与许多内源性和外源性化合物结合,是一种重要的存储和转运蛋白。

简述药物和血浆蛋白结合的特点。
药物和蛋白质的结合是可逆的,结合的程度取决于药物的亲和力和血浆蛋白的含量。
一般来说,药物的亲和力越强,结合的程度越高。
结合后的药物分子无法通过细胞膜进行扩散,因此无法发挥药效,只有游离的药物分子才能对靶细胞产生作用。
药物和血浆蛋白的结合还可以影响药物的药代动力学和药效学,包括药物的半衰期、清除率、毒性等。
此外,药物和蛋白质结合还可能引起药物相互作用,从而干扰药物的疗效或增加药物的毒副作用。
因此,在药物研发和药物治疗中,了解药物和血浆蛋白结合的特点及其影响是非常重要的。
- 1 -。

血浆蛋白结合率名词解释药理学嘿,你知道啥是血浆蛋白结合率不?这在药理学里可是个相当重要的概念呢!血浆蛋白结合率呀,就好比是一场舞会里,药物分子和血浆蛋白之间的“亲密互动”比例。
比如说,药物就像是一个急切想找到舞伴的人,而血浆蛋白就是那些受欢迎的舞伴啦!
有些药物特别“抢手”,能和血浆蛋白紧密结合,结合率就高;而有些药物可能就没那么受欢迎,结合率就低喽。
这就好像有的人在舞会上能快速找到心仪的舞伴,而有的人却只能在旁边干瞪眼。
你想想看啊,要是一种药物的血浆蛋白结合率很高,那它在血液里大部分时间都和血浆蛋白“黏”在一起,能发挥作用的游离药物就少了呀!这就像你和好朋友手牵手,那你就没办法同时去做其他事情啦。
咱再举个例子哈,有一种药叫阿司匹林,它的血浆蛋白结合率不算特别高。
这意味着啥呢?意味着有相当一部分阿司匹林是可以自由活动的,能更快地去发挥它的药效,去缓解疼痛、消炎啥的。
那血浆蛋白结合率对药物的作用有啥影响呢?这可太重要啦!它会影响药物的分布、代谢和排泄呢!就像你参加完舞会,你的行动和后续安排都会受到你舞伴的影响一样。
如果结合率高,药物在体内的停留时间可能就长,药效持续时间也长;但如果结合率低,可能药效就没那么持久啦。
我觉得吧,血浆蛋白结合率就像是药物世界里的一个神秘密码,解开了它,我们就能更好地理解药物的行为和效果。
所以呀,搞清楚血浆蛋白结合率,对我们正确用药、理解药物作用可太重要啦!你说是不是呢?反正我是这么认为的!。

降低血浆蛋白结合的结构修饰策略降低血浆蛋白结合的结构修饰策略自从人类认识到药物与血浆蛋白的结合程度会直接影响药物的药效和代谢过程后,降低蛋白结合率成为新药研发领域的一个重要课题。
为了降低药物与血浆蛋白的结合,许多结构修饰策略被提出并不断地被研究和优化,其中包括以下几种:1. 脂溶性降低策略:脂溶性是影响药物与血浆蛋白结合率的一个重要因素。
通过在分子结构中引入极性基团,使药物分子变得更亲水,可以降低其与血浆蛋白的结合。
例如,羧酸和磺酸基团可以增加药物分子的极性,降低其脂溶性,从而降低药物与血浆蛋白的结合率。
2. 酸碱度调控策略:药物分子的酸碱度也是影响与血浆蛋白结合的重要因素。
通过调节药物分子的酸碱度,可以改变其与血浆蛋白结合的程度。
例如,药物分子中的酸性基团可以与血浆蛋白中的碱性残基结合,从而降低药物与血浆蛋白的结合率。
3. 母体激肽修饰策略:母体激肽修饰是一种通过改变药物分子中的特定氨基酸残基,从而改变其与血浆蛋白结合的策略。
例如,将药物分子中的酪氨酸残基改变为酪氨酰硒酸酯,可以使药物与血浆蛋白结合的亲和力降低。
4. 选择性配体修饰策略:选择性配体修饰是一种通过在药物分子中引入特定的配体,使其与血浆蛋白结合的部位发生改变,从而降低药物与血浆蛋白结合的程度。
例如,通过引入硫醇基团,使药物与血浆蛋白中的巯基结合,从而降低其与其他位点的结合。
5. 多基团调控策略:多基团调控是一种通过在药物分子中引入多个基团,使其与血浆蛋白结合的模式发生改变,从而降低药物与血浆蛋白的结合率。
例如,通过引入两个磺酸基团,使药物与血浆蛋白的结合形成一个桥接结构,从而降低药物与血浆蛋白的结合。
综上所述,通过脂溶性降低策略、酸碱度调控策略、母体激肽修饰策略、选择性配体修饰策略和多基团调控策略等多种结构修饰策略,可以有效降低药物与血浆蛋白的结合,提高药物的药效和代谢效率。
这些策略的不断研究和改进将为新药的开发和临床应用提供更多的选择和可能性,为人类的健康事业做出更大的贡献。