药物与血浆蛋白结合
- 格式:docx
- 大小:17.06 KB
- 文档页数:2

药物的血浆蛋白结合是指药物进入循环后首先与血浆蛋白结合,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,联合药物的药理活性暂时消失。
当分子膨胀时,结合物不能通过毛细血管壁,所以它们可以暂时“储存”在血液中。
药物与血浆蛋白结合的特异性较低,但其结合位点有限。
两种药物可能与同一种蛋白质竞争替代品。
例如,蛋白酶与双香豆素竞争血浆蛋白,这会增加血浆中的自由形式浓度,从而可能导致出血。
这种组合的特点如下可逆性结合后,药物活性暂时消失:结合物变大,不能通过毛细血管壁暂时储存在血液中,不经分布和消除。
可能发生竞争性取代:药物与血浆蛋白结合的特异性较低,但血浆蛋白结合位点有限。
两种药物可能与同一种蛋白质竞争产生替代品。
结合率如下药物的血浆蛋白结合能力受药物浓度、血浆蛋白质量和数量、解离常数的影响。
药理学书籍记载的药物血浆蛋白结合率是正常人在正常剂量范围内测得的值。
影响药物血浆蛋白结合率的因素和后果如下当两种药物同时使用时,会发生竞争性替代。
如果一种药物的结合率达到99%,就会被另一种药物取代,降低1%,理论上游离(药理活性)药物的浓度将增加100%,可能导致中毒。
然而,在游离药物不断增加的过程中,血浆中的药物浓度会不断增加。
药物也可能与内源性代谢物竞争并与血浆蛋白结合,如磺胺类药物交换胆红素和血浆蛋白结合,这可能导致新生儿核黄疸。
当血浆蛋白过低(如肝硬化)或恶化(如尿毒症)时,药物的血浆蛋白结合率降低,容易引起毒性反应。
由于血浆蛋白有限,当结合率高的药物在结合部位达到饱和时,如果药物剂量继续增加,血浆中的游离药物浓度将大大增加,引起毒性反应。


1药物与血浆蛋白结合(c)。
A是不可逆的B.加速药物在体内的分布C.是疏松和可逆的D.促进药物排泄E.无饱和性和臵换现象2某弱酸性药在pH为5时约90%解离,其pKa值为(c)。
A.6 B.5C.4D.3E.23吸收最慢的是(d)。
A.口服给药B.静脉注射C.舌下含服D.经皮给药E.吸入给药4主动转运的特点是(a)。
A.需要载体,消耗能量B.需要载体,不消耗能量C.消耗能量,无饱和性D.无饱和性,有竞争性抑制E.不消耗能量,无竞争性抑制5对消除半衰期的认识不正确的是(b)。
A.药物的血浆浓度下降一半所需的时间B.药物的组织浓度下降一半所需的时间C.临床上常用消除半衰期来反映药物消除的快慢D.符合零级动力学消除的药物,其半衰期与体内药量有关E.一次给药后,经过5个半衰期体内药物已基本消除使激动剂的量效曲线平行右移,最大效应不变的是(e)。
A.拮抗剂B.激动剂C.部分激动剂D.非竞争性拮抗剂E.竞争性拮抗剂某药的半衰期为10小时,一次给药后从体内基本消除的时间是(a)。
A.约50小时B.约30小时C.约80小时D.约20小时E.约70小时血药浓度已降到有效浓度以下时为(d)。
A.潜伏期B.持续期C.失效期D.残留期E.消除半衰期与受体有亲和力但无内在活性的是(a)。
A.拮抗剂B.激动剂C.部分激动剂D.非竞争性拮抗剂E.竞争性拮抗剂血药浓度维持在最低有效浓度之上的时间是(b)。
A.潜伏期B.持续期C.失效期D.残留期E.消除半衰期副作用是在哪种剂量下产生的不良反应(b)。
A.最小有效量B.治疗剂量C.大剂量D.阈剂量E.与剂量无关消除半衰期的长短取决于(b)。
A.药物的吸收速率B.药物的消除速率C.药物的转化速率D.药物的转运速度E.药物的分布速率长期使用肾上腺皮质激素停药后,表现为肾上腺皮质功能低下属于(d)。
A.变态反应B.特异质反应C.停药反应D.后遗效应E.快速耐受性可能有首关消除的是(a)。


药物的血浆蛋白结合是指药物进入循环后首先与血浆蛋白结合,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,结合药物的药理活性暂时消失。
由于分子扩大,结合物不能通过毛细血管壁,因此它们可以暂时“储存”在血液中。
药物与血浆蛋白结合的特异性低,但是血浆蛋白的结合位点受到限制。
两种药物可能会与同一种蛋白质竞争替代品。
例如,宝泰松与双香豆素竞争血浆蛋白,这会增加血浆中的游离型浓度,这可能导致出血。
组合的特征如下可逆性结合后,药理活性暂时消失:结合物变大,无法通过毛细血管壁暂时储存在血液中,而没有分布和消除。
可能发生竞争性替代:药物与血浆蛋白结合的特异性低,但血浆蛋白的结合点有限。
两种药物可能会与同一蛋白质竞争而产生替代现象。
结合率如下药物的血浆蛋白结合能力受药物浓度,血浆蛋白的质量和数量以及解离常数的影响。
药理学书籍中记录的药物血浆蛋白结合率是在正常剂量范围内对正常人测得的值。
影响药物血浆蛋白结合率的因素及后果如下当两种药物一起使用时,就会发生竞争性替代。
如果一种药物的结合率达到99%,则当其被另一种药物替代并降低1%时,理论上游离型(具有药理活性)药物的浓度将增加100%,这可能导致中毒。
但是,在普通药物的更换过程中,游离药物将被迅速清除,血浆中游离药物的浓度难以持续增加。
药物也可能与内源性代谢物竞争并与血浆蛋白结合,例如磺酰胺交换胆红素和血浆蛋白结合,这可能导致新生儿发生核黄疸。
当血浆蛋白过少(如肝硬化)或变质(如尿毒症)时,药物血浆蛋白的结合率降低,容易引起毒性反应。
由于血浆蛋白有限,当具有高结合率的药物在结合位点达到饱和时,如果药物剂量继续增加,血浆中游离药物的浓度将大大增加并引起毒性反应。


药物的血浆蛋白结合是指药物进入循环后首先与血浆蛋白结合,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,联合药物的药理活性暂时消失。
当分子膨胀时,结合物不能通过毛细血管壁,所以它们可以暂时“储存”在血液中。
药物与血浆蛋白结合的特异性较低,但血浆蛋白结合位点有限。
两种药物可能与同一种蛋白质竞争替代品。
例如,蛋白酶与双香豆素竞争血浆蛋白,这会增加血浆中的自由形式浓度,从而可能导致出血。
这种组合的特点如下可逆性结合后,药物活性暂时消失:结合物变大,不能通过毛细血管壁暂时储存在血液中,不经分布和消除。
可能发生竞争性取代:药物与血浆蛋白结合的特异性较低,但血浆蛋白的结合位点有限。
两种药物可能与同一种蛋白质竞争产生替代物。
结合率如下药物的血浆蛋白结合能力受药物浓度、血浆蛋白质量和数量以及解离常数的影响。
药理学书籍记载的药物血浆蛋白结合率是正常人在正常剂量范围内测得的值。
影响药物血浆蛋白结合率的因素和后果如下当两种药物同时使用时,会发生竞争性替代。
如果一种药物的结合率达到99%,被另一种药物取代并降低1%,理论上游离(药理活性)药物的浓度将增加100%,可能导致中毒。
但在更换常用药物的过程中,游离药物会迅速被清除,血浆中游离药物浓度难以持续升高。
药物也可能与内源性代谢物竞争并与血浆蛋白结合,如磺胺类药物交换胆红素和血浆蛋白结合,这可能导致新生儿核黄疸。
当血浆蛋白过低(如肝硬化)或恶化(如尿毒症)时,药物血浆蛋白结合率降低,容易引起毒性反应。
由于血浆蛋白有限,当结合率高的药物在结合部位达到饱和时,如果药物剂量继续增加,血浆中游离药物浓度将大大增加,引起毒性反应。

药物与血浆蛋白结合该药物的血浆蛋白结合情况如下药物进入循环后,首先成为与血浆蛋白结合的药物,未结合的药物称为游离药物。
药物与血浆蛋白的结合是可逆的,偶联药物的药理活性暂时消失。
由于分子的膨胀,结合物不能通过毛细血管壁,所以它们可以暂时“储存”在血液中。
药物与血浆蛋白的结合特异性较低,但血浆蛋白的结合位点有限。
两种药物可能会与同一种蛋白质争夺替代品。
如保泰松与双香豆素争夺血浆蛋白,增加了后者的游离型浓度,可能导致出血。
结合特点:可逆结合后药理活性暂时消失;结合物变大,不能通过毛细血管壁暂时储存在血液中,不能分布和消除。
可能出现竞争性替代:药物与血浆蛋白结合的特异性较低,但血浆蛋白结合点有限。
两种药物可能与同一种蛋白质竞争,产生替代现象。
结合率如下药物浓度、血浆蛋白的质量和数量以及解离常数影响药物的血浆蛋白结合能力。
药理学书籍中记录的药物血浆蛋白结合率是正常人群在常用剂量范围内测量的值。
影响药物血浆蛋白结合率的因素及后果如下当两种药物同时使用时,出现竞争性替代。
如果一种药物的结合率达到99%,被另一种药物取代后降低1%,理论上游离型(具有药理活性)药物的浓度就会增加100%,可能导致中毒。
然而,在普通药物被替代的过程中,游离药物会迅速被淘汰,血浆中游离药物的浓度难以持续升高。
药物也可能与内源性代谢物与血浆蛋白结合,如磺胺类药交换胆红素和血浆蛋白结合,这可能会导致新生儿核黄疸。
当血浆蛋白过少(如肝硬化)或病情恶化(如尿毒症)时,药物血浆蛋白结合率降低,容易引起毒性反应。
由于血浆蛋白的限制,当结合率高的药物在结合位点达到饱和时,如果药物剂量继续增加,血浆中游离药物的浓度会大大增加,引起毒性反应。

药物与血浆蛋白结合的药理学研究对于药物和血浆蛋白的结合,通常的认知是二者形成的复合体是不能够实现跨膜运转的,进而使机体摄入的药物在分布、代谢、排泄及与相应的受体在结合后产生的药理效果,会以一种游离的形式进行,而游离药物在血液中发生的浓度变化是对机体内药物处置、药效起到决定性影响的因素中的重要一种。
本文对药物与血浆蛋白结合的药理学基础和研究进展进行阐述,对临床的常规用药需要考虑的因素进行总结,从而能够明确药物在何种情况下需要监测游离的浓度。
1药物与血浆蛋白的结合机体在使用药物后,药物进入机体内循环,由于结构上的差异性,就会与血细胞、血浆蛋白互相结合,形成结合型药物,而没有发生结合的药物,被称为游离药物。
药物在不同的作用下进行结合,这些作用分别是共价键结合、离子键吸引、氢键结合、电荷转移、疏水性结合及范德华引力,而上述的药物结合方式是可逆的,如果结合后,药物分子发生变大的情况,就不易透膜,能够在血液中储存,然后经过血液运输,通过游离药物分布到机体的各个组织部位,进而起到治疗的作用。
在血液中,多数药物能够与血浆蛋白结合,能够与血细胞结合,或者进入血细胞,当进入血细胞后,分布在血液中,继而形成了一种动态的平衡。
但在上述的各种结合方式中,药物与血浆蛋白的结合会对药物的分布造成重要的影响,特别是药物结合成的蛋白组分中,白蛋白(HSA)和α1酸性糖蛋白(AGP)是2种最为重要的。
1.1结合方式为保证药物的安全性和临床用药的有效性,人们在研究中,将药物与血浆蛋白的结合规律进行了重点研究,研究发现,药物与血浆蛋白的结合方式,对患者体内的药物浓度起到预测的作用,根据质量守恒的作用原理,药物和血浆蛋白的可逆结合要保证以下的平衡:ka=k1/k-1=1/kd(1)式(1)中,k1是结合速率常数,解离速率常数是k-1,药物-蛋白复合物结合常数是ka,解离常数是kd。
该公式反应的是药物与蛋白在结合产生的亲和力的大小。
高蛋白与药物结合的ka值范围在105~107mmol/L,而低蛋白结合的ka值和中等结合强度ka的值范围均在102~104mmol/L。
药物与血浆蛋白结合的药理学研究对于药物和血浆蛋白的结合,通常的认知是二者形成的复合体是不能够实现跨膜运转的,进而使机体摄入的药物在分布、代谢、排泄及与相应的受体在结合后产生的药理效果,会以一种游离的形式进行,而游离药物在血液中发生的浓度变化是对机体内药物处置、药效起到决定性影响的因素中的重要一种。
本文对药物与血浆蛋白结合的药理学基础和研究进展进行阐述,对临床的常规用药需要考虑的因素进行总结,从而能够明确药物在何种情况下需要监测游离的浓度。
1药物与血浆蛋白的结合机体在使用药物后,药物进入机体内循环,由于结构上的差异性,就会与血细胞、血浆蛋白互相结合,形成结合型药物,而没有发生结合的药物,被称为游离药物。
药物在不同的作用下进行结合,这些作用分别是共价键结合、离子键吸引、氢键结合、电荷转移、疏水性结合及范德华引力,而上述的药物结合方式是可逆的,如果结合后,药物分子发生变大的情况,就不易透膜,能够在血液中储存,然后经过血液运输,通过游离药物分布到机体的各个组织部位,进而起到治疗的作用。
在血液中,多数药物能够与血浆蛋白结合,能够与血细胞结合,或者进入血细胞,当进入血细胞后,分布在血液中,继而形成了一种动态的平衡。
但在上述的各种结合方式中,药物与血浆蛋白的结合会对药物的分布造成重要的影响,特别是药物结合成的蛋白组分中,白蛋白(HSA)和α1酸性糖蛋白(AGP)是2种最为重要的。
1.1结合方式为保证药物的安全性和临床用药的有效性,人们在研究中,将药物与血浆蛋白的结合规律进行了重点研究,研究发现,药物与血浆蛋白的结合方式,对患者体内的药物浓度起到预测的作用,根据质量守恒的作用原理,药物和血浆蛋白的可逆结合要保证以下的平衡:ka=k1/k-1=1/kd(1)式(1)中,k1是结合速率常数,解离速率常数是k-1,药物-蛋白复合物结合常数是ka,解离常数是kd。
该公式反应的是药物与蛋白在结合产生的亲和力的大小。
高蛋白与药物结合的ka值范围在105~107mmol/L,而低蛋白结合的ka值和中等结合强度ka的值范围均在102~104mmol/L。

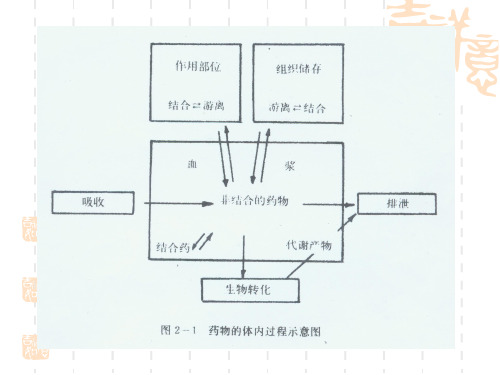
药学——临床药学药理学(药剂专业掌握)单选题(63题,共63分)1.药物在血浆中与血浆蛋白结合后,下列正确的是(1分)A.药物作用增强B.药物代谢加快C.药物排泄加快D.暂时失去药理活性E.药物转运加快正确答案:D本题分数:1分答案解析:药物与血浆蛋白结合,属于结合性药物,类似药库的作用。
特点即为暂时失去药理活性,体积增大,不易通过血管壁,暂时“储存”于血液中。
2.普鲁卡因青霉素之所以能长效,是因为(1分)A.改变了青霉素的化学结构B.抑制排泄C.减慢了吸收D.延缓分解E.加进了增效剂正确答案:C本题分数:1分答案解析:因为普鲁卡因是一种麻醉剂,它可以使血管收缩,组织渗出减少,从而阻碍了青霉素的吸收,所以普鲁卡因青霉素肌注后吸收缓慢。
知识点1:药物的体内过程知识点2:排泄难度:13.以下适用于治疗支原体肺炎的是(1分)A.庆大霉素B.两性霉素BC.氨苄西林D.四环素E.氯霉素正确答案:D本题分数:1分答案解析:该品为广谱抑菌剂,高浓度时具杀菌作用。
除了常见的革兰氏阳性菌、革兰氏阴性菌以及厌氧菌外,多数立克次体属、支原体属、衣原体属、非典型分枝杆菌属、螺旋体也对该品敏感。
知识点1:抗菌药的临床应用知识点2:四环素类难度:14.有关喹诺酮类性质和用途的叙述,错误的是(1分)A.萘啶酸为本类药物的第一代,仅用于尿路感染,其作用弱B.吡哌酸对尿路感染有效外,用于肠道感染及中耳炎C.第三代该类药物的分子中均含氟原子D.本类药物可以代替青霉素G用于上呼吸道感染E.环丙沙星属于第三代喹诺酮类抗菌药物正确答案:D本题分数:1分答案解析:喹诺酮类是主要作用于革兰阴性菌的抗菌药物,对革兰阳性菌的作用较弱(某些品种对金黄色葡萄球菌有较好的抗菌作用)。
上呼吸道感染主要是由病毒引起的,因此,其不能代替青霉素G的作用。
知识点1:抗菌药的临床应用知识点2:合成抗生素难度:25.应用乙醚麻醉前给予阿托品,其目的是(1分)A.协助松弛骨骼肌B.防止休克C.解除胃肠道痉挛D.减少呼吸道腺体分泌E.镇静作用正确答案:D本题分数:1分答案解析:阿托品为阻断M胆碱受体的抗胆碱药,能解除平滑肌的痉挛(包括解除血管痉挛,改善微血管循环);抑制腺体分泌;解除迷走神经对心脏的抑制,使心跳加快;散大瞳孔,使眼压升高;兴奋呼吸中枢。

药物与血浆蛋白结合药物与血浆蛋白结合率的变化通过影响游离型药物浓度,改变药物分布、代谢、排泄以及作用靶点的结合,从而影响药理效应及毒副作用。
对1500种常用药的研究表明,43%化合物的血浆蛋白结合大于90%。
血浆蛋白结合在不同的治疗领域没有显著差异:中枢神经系统、炎症和肾-心血管。
唯一的例外可能是抗炎药物,其高蛋白结合者(>99%)占较高的比例(26%),且其中大多数为酸类。
令人惊奇的是,许多中枢神经系统药物具有高血浆蛋白结合的特点。
该研究最惊人的发现是,化学治疗药物(包括抗生素、抗病毒药物、抗真菌药物和抗肿瘤药物)中有较高比例(77%)的低结合药物。
在设计化学治疗药物方面,低血浆蛋白结合似乎更有优势。
血浆蛋白结合的基本原理血液经抗凝处理后的全部血液为全血;血浆是血液(不包括细胞)的液体部分。
在存在抗凝剂(如肝素)的条件下采集新鲜血液,然后离心去除血细胞而得到用于体外研究的血浆,血浆蛋白仍然保留于液体部分。
血清是去除了凝血因子(如纤维蛋内原)的血浆,它是在不含抗凝剂条件下采集的。
药动学研究通常采集血浆,从而保留了蛋白结合型及游离型药物,但除去了与细胞结合的药物。
许多药物与血浆蛋白、组织蛋白或体内的大分子物质如白蛋白、DNA 等反应,生成药物大分子复合物。
药物与蛋白类高分子结合后,分子体积变大,不能透过血管壁向组织转运,不能由肾小球滤过,不能透过胎盘屏障,不能经肝代谢。
进人血液中的药物,一部分在血液中呈游离形式存在,一部分与血浆蛋白通过离子键、氢键、疏水键及范德华力可逆性结合,形成药物血浆蛋白复合物,是药物在血浆中的一种贮存形式,能降低药物的分布与消除速度,维持药物疗效。
药物的蛋白结合不仅影响药物的体内分布,而且还影响药物的代谢和排泄。
药物在机体内最重要的结合就是蛋白结合,且主要是血浆蛋白结合。


药物与血浆蛋白结合
血液经抗凝处理后的全部血液为全血;
血浆是血液(不包括细胞)的液体部分。
在存在抗凝剂(如肝素)的条件下采集新鲜血液,然后离心去除血细胞而得到用于体外研究的血浆,血浆蛋白仍然保留于液体部分。
血清是去除了凝血因子(如纤维蛋内原)的血浆,它是在不含抗凝剂条件下采集的。
药动学研究通常采集血浆,从而保留了蛋白结合型及游离型药物,但除去了与细胞结合的药物。
许多药物与血浆蛋白、组织蛋白或体内的大分子物质如白蛋白、DNA 等反应,生成药物大分子复合物。
药物与蛋白类高分子结合后,分子体积变大,不能透过血管壁向组织转运,不能由肾小球滤过,不能透过胎盘屏障,不能经肝代谢。
进人血液中的药物,一部分在血液中呈游离形式存在,一部分与血浆蛋白通过离子键、氢键、疏水键及范德华力可逆性结合,形成药物血
浆蛋白复合物,是药物在血浆中的一种贮存形式,能降低药物的分布与消除速度,维持药物疗效。
药物的蛋白结合不仅影响药物的体内分布,而且还影响药物的代谢和排泄。
药物在机体内最重要的结合就是蛋白结合,且主要是血浆蛋白结合。
药物的血浆蛋白结合率是影响药物分布的重要因素。
血液中结合型的药物不易透过细胞膜,只有游离型的药物可向血管外扩散,转运到组织中。
如果药物的血浆蛋白结合率高,血浆中游离型药物浓度少,进入其他组织的浓度就低。
因此,药物分布主要取决于血液中的游离型药物的浓度,另外也与该药物和组织结合程度有很大关系。
蛋白结合对药物分布的影响见表4-5,可见血浆中游离型药物浓度越高,越容易向其他组织转运,药物的表观分布容积越大。

对药物与血浆蛋白结合后的描述哪项有错误药物与血浆蛋白结合是指药物在体内与血浆蛋白相互结合,并形成药物-蛋白复合物的过程。
这个过程对于药物的分布、代谢和排泄都有重要影响。
在描述药物与血浆蛋白结合后的情况时,需要注意以下几个方面。
首先,要了解药物与血浆蛋白结合的特点。
药物与血浆蛋白结合的特点有选择性、可逆性和饱和性。
选择性是指不同药物对不同的蛋白质有不同的亲和力,药物通常会选择性地结合于血浆中的白蛋白。
可逆性是指药物与蛋白质结合的过程可以反复进行,而且结合的药物可以与蛋白质分离。
饱和性是指当血浆中的药物浓度足够高时,所有能结合的蛋白位点都被占满,这时进一步增加药物剂量无法增加结合率。
其次,要正确描述药物与血浆蛋白结合的影响。
药物与血浆蛋白结合后,只有游离态的药物才能发挥药效,因此结合率越高,有效药物浓度就越低。
结合率高的药物需要更高的剂量才能达到治疗效果,这也增加了药物的不良反应的风险。
另外,药物与血浆蛋白结合后,药物在体内的分布受到限制,无法自由穿过细胞膜进入组织或穿过血脑屏障进入中枢神经系统,从而影响了药物的疗效。
最后,要明确描述哪项有错误。
根据我给出的标题,对药物与血浆蛋白结合后的描述中,可能存在的错误是指述药物与蛋白质结合为一种不可逆的过程。
实际上,药物与蛋白质的结合是一个可逆的过程,药物与蛋白质间通过非共价键结合,这种结合可以在体内自发发生,也可以通过竞争性结合的药物来解离,使药物回到游离状态。
因此,结合的药物不是永久不可逆的。
总结起来,描述药物与血浆蛋白结合后的情况时,应该准确说明药物与蛋白质结合的特点,以及结合对药物的药效、副作用和分布的影响。
同时,需要确保描述中的内容准确无误,以避免引起误解。

血浆是一种水溶液,包含92%的水,7%的蛋白质以及1%的其它物质(比如无机盐),总共占到人体血液的55%的组分。
药物与血浆蛋白的结合(Plasma Protein Binding, PPB)会降低血液循环系统中自由药物浓度,从而影响渗透到组织细胞到达药物靶点的治疗能力,或者影响肾脏的消毒能力。
因此,PPB的结合事件会影响药物在体内的活性代谢时间和毒副作用,患者的其他药物,食物和病理状况的共同给药可以显着改变药物的结合百分比,并可能导致严重后果。
药物分子可以通过多种方式与血浆蛋白结合,而且血浆蛋白结合作用是可逆的,主要取决于疏水作用和静电作用,比如vdW作用和氢键。
血浆蛋白结合药物与游离态药物的浓度达成动态平衡,这个可逆平衡过程会强烈影响各种药理学性质,比如分布体积,药物清除率,和消除,以及药理效应。
因为只有一部分游离药物可以跨过细胞膜,高蛋白结合能力的药物比低蛋白结合能力的药物有更长的半衰期。
结合到血浆蛋白上的药物越多,发挥治疗作用的游离药物的比例越小。
血浆蛋白对药物的结合能力是评估药动力学特点的一个非常重要的性质,评估游离药物在组织器官和血液中的比例是药物设计中的一个有价值的科学问题。
血浆蛋白主要包括白蛋白、球蛋白、凝血因子和调节蛋白,最重要的药物结合蛋白是白蛋白和α1-酸性糖蛋白,其次是脂蛋白。
其中,白蛋白的浓度最高(600μM),是血浆中的主要药物结合组分(人类血清白蛋白,HSA,human serum albumin);其次是α-酸性糖蛋白(AAG,α-acid glycoprotein,α-也叫做乳清类粘蛋白),12-30μM,和脂蛋白(lipoproteins,也叫做γ-球蛋白)。
关于药物结合HSA和AAG的研究工作是最多的。
白蛋白是人类血浆蛋白中的主体组分(600 μM),含量占到总血浆蛋白的60%。
白蛋白(HSA),是人血浆中含量最丰富的一种蛋白质,能与许多内源性和外源性化合物结合,是一种重要的存储和转运蛋白。
药物与血浆蛋白结合具有( )。
A.可逆性
B.不可逆性
C.竞争性抑制现象
D.竞争置换现象
E.饱和性
解析:药物的血浆蛋白结合指的是药物进入循环后首先与血浆蛋白成为结合型药物,未被结合的药物称为游离型药物。
药物与血浆蛋白的结合是可逆的,结合型药物的药理活性暂时消失,结合物因分子变大不能通过毛细血管壁而暂时“储存”于血液中。
药物与血浆蛋白结合特异性低,而血浆蛋白结合位点有限,两个药物可能竞争与同一蛋白结合而发生置换现象,例如保泰松与双香豆素竞争血浆蛋白,使后者游离型浓度增高,可导致出血。
结合的特点:
可逆性
结合后药理活性暂时消失:结合物分子变大不能通过毛细管壁暂时“储存”于血液中,不进行分布和消除。
可发生竞争置换:药物与血浆蛋白结合特异性低,而血浆蛋白结合点有限,两个药物可能竞争与同一蛋白结合而发生置换现象。
结合率:
药物的血浆蛋白结合量受药物浓度,血浆蛋白的质和量及解离常数的影响,各药不同而且结合率随剂量增大而减少。
药理学书籍收载的药物血浆蛋白结合率是在常用剂量范围内对正常人测定的数值。
影响药物血浆蛋白结合率的因素及后果:
两药合用时发生竞争置换,如某药结合率达99%,当被另药置换而下降1%时,则游离型(具有药理活性)药物浓度在理论上将增加100%,可能导致中毒。
但一般药物在被置换过程中,游离型药物会加速被消除,血浆中游离型药物浓度难以持续增高。
药物也可能与内源性代谢物竞争与血浆蛋白结合,如磺胺药置换胆红素与血浆蛋白结合,在新生儿可能导致核黄疸症。
血浆蛋白过少(如肝硬化)或变质(如尿毒症)时药物血浆蛋白结合率下降,容易发生毒性反应。
由于血浆蛋白有限,当结合率高的药物在结合部位达到饱和后,如继续增加药量,将导致血浆中游离型药物浓度大增,引起毒性反应。