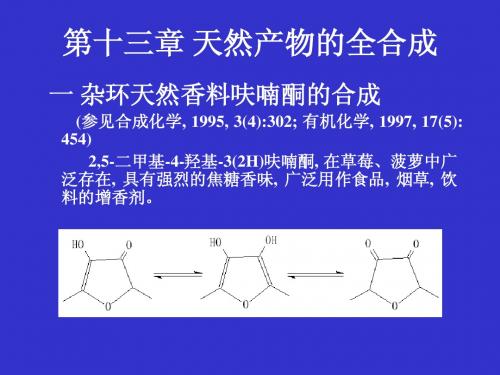
天然产物全合成实例
- 格式:ppt
- 大小:202.50 KB
- 文档页数:36

天然产物的全合成方法研究天然产物是指存在于自然界中的有机化合物,如植物、动物和微生物产生的化学物质。
这些化合物通常具有复杂的结构和多样的生物活性,因此对于天然产物的全合成方法的研究具有重要的科学意义和应用价值。
天然产物的全合成方法是指通过人工合成的方式,将天然产物的结构完全复制出来。
在过去的几十年里,许多科学家致力于发展新的合成方法,以便能够高效地合成天然产物。
这些合成方法涉及到有机合成化学的各个方面,包括反应的选择性、底物的设计和合成路径的优化等。
在天然产物的全合成方法研究中,反应的选择性是一个非常重要的考虑因素。
天然产物的结构通常包含多个官能团,因此在合成过程中需要选择适当的反应条件,以确保特定官能团的保护和反应的进行。
例如,醛和酮等羰基化合物在合成过程中往往需要选择适当的还原剂或氧化剂来进行反应。
此外,选择性的控制还需要考虑到其他官能团的稳定性和反应性,以避免不必要的副反应的发生。
底物的设计也是天然产物全合成方法研究中的一个重要方面。
由于天然产物的结构复杂多样,因此在设计底物时需要考虑到合成路径的可行性和效率。
有时候,为了合成一个复杂的天然产物,需要利用多步反应和中间体的转化。
在这种情况下,底物的设计需要考虑到每一步反应的可行性和产物的稳定性。
此外,底物的设计还需要考虑到合成过程中可能产生的副产物和废弃物,以确保合成方法的可持续性和环境友好性。
合成路径的优化是天然产物全合成方法研究中的另一个重要方面。
在合成过程中,通常需要通过多步反应来构建复杂的结构。
因此,合成路径的优化需要考虑到每一步反应的条件和反应物的选择,以最大程度地提高合成的效率和产物的纯度。
此外,合成路径的优化还需要考虑到反应物和中间体的稳定性,以避免不必要的副反应和产物的分解。
天然产物的全合成方法研究不仅对于理论化学有着重要的意义,也对于药物研发和生物活性研究具有重要的应用价值。
通过合成天然产物,可以获得足够的化合物量,以进行药物活性的评价和生物学机制的研究。


药用天然产物全合成 - 合成路线精选在当今社会,人们对健康的重视程度日益提高,对药用天然产物的需求也越来越大。
药用天然产物是指从天然植物、动物或微生物中提取的具有药用价值的化合物,其来源自然、含量丰富、效果明显。
然而,由于天然产物的含量一般较低,提取困难,以及在纯度、稳定性和效力方面存在局限性,因此人们对药用天然产物全合成的研究日益增多。
在全合成药用天然产物的研究领域,合成路线的设计和精选显得尤为重要。
合成路线的设计涉及到从简化的原料开始,经过多步反应,最终得到目标化合物的过程。
优秀的合成路线应当具有高效性、高收率、低成本、稳定可控、环境友好等特点。
在选择合成路线时,应当优先考虑以下几个方面:1. 定位于目标化合物的结构特点,以及结构改造的难易程度。
2. 合成途径的可行性和可操作性。
3. 合成途径所需试剂和条件的可获取性和可操作性,是否会引入有毒、难处理的中间体或副产物。
4. 合成途径的收率、原子效率以及废弃物处理。
接下来,我将通过从简化的原料开始,逐步进行深入研究,为您介绍某种主题的合成路线。
第一步:从某种简化的原料出发,进行反应A,得到中间体X。
中间体X具有一定反应活性,容易进行进一步的转化和改造。
第二步:对中间体X进行反应B,得到中间体Y。
在该步骤中,需特别注意反应条件的控制,以避免副反应的发生,并保证高收率。
第三步:对中间体Y进行反应C,得到目标化合物Z。
在该步骤中,可以选择不同的合成途径,以达到最佳的合成效果。
经过以上步骤的设计和优化,得到了一个高效、高收率、低成本的合成路线,从而成功地合成了目标化合物Z。
这种合成路线的设计和精选,将为药用天然产物全合成提供重要的范例和启发。
个人观点和理解:在进行药用天然产物全合成研究时,合成路线的设计和精选非常重要。
合成路线的选择应当以简化的原料为出发点,以高效、高收率、低成本为目标,经过多次反应得到目标化合物。
还应当考虑到合成途径的可行性、可操作性、废弃物处理等方面。


2008年第28卷有机化学V ol. 28, 2008第5期, 918~921 Chinese Journal of Organic Chemistry No. 5, 918~921yang_jh@*E-mail:Received September 22, 2007; revised November 3, 2007; accepted December 28, 2007.No. 5杨金会等:天然产物1,3-二-(2-羟基-4-甲氧基苯基)丙烷和1,3-二-(2,4-二羟基苯基)丙烷的合成9191,3-二-(2-羟基-4-甲氧基苯基)丙烷(1)和1,3-二-(2,4-二羟基苯基)丙烷(2)是从菊叶薯蓣(Dioscorea composita Hemsl.)的干燥根茎中分离出来的[7], 如Scheme 1, 它们的合成尚未见文献报道. 菊叶薯蓣(Dioscorea composita Hemsl.)原产于墨西哥, 1978年在云南的西双版纳开始种植, 是合成类固醇激素的薯蓣皂苷配基的主要来源[7].Scheme 1以间苯二酚作为起始原料, 经羰基化、选择性的甲基化、酚羟基保护、缩合、还原等步骤, 首次完成了化合物1和2的全合成. 合成路线如Scheme 2所示.Reagents and conditions: (a) CH 3COOH/ZnCl 2, 76%; (b) Me 2SO 4, K 2CO 3, acetone, 83%; (c) DMF, POCl 3, 77%; (d) Me 2SO 4, K 2CO 3, acetone, 81%; (e) ClMOM, KOH, (C 4H 9)NI, CH 2Cl 2/H 2O, 98%; (f) KOH, EtOH-H 2O, 0℃~r.t., 36 h, 81%; (g) H 2, Pd/C, EtOAc, 0 ℃, 3 h, 81%; (h) Zn/Hg, HCl, H 2O, toluene, r.t., 74%; (i) BBr 3, CH 2Cl 2, 85%.Scheme 21 实验部分1.1 仪器与实验熔点在XT 4-100x 熔点仪上测定, 熔点仪未校正; 红外光谱在Nicolet AVATAR 360 FT-IR 型红外光谱仪上测定; 1H NMR 数据在Burker AM-400测定, 用CDCl 3或CD 3OD 作溶剂, TMS 作内标; 质谱数据在ZAB-HS 型质谱仪上测定. 柱层析所用硅胶均为青岛海洋化工厂产品(200~300目).1.2 2,4-二羟基苯乙酮(4)的合成取14.0 g (100 mmol)无水ZnCl 2, 30 mL 冰醋酸, 密封搅拌加热使无水ZnCl 2完全溶解. 快速分批加入11.00 g (100 mmol) 1,3-二羟基苯酚, 回流反应1 h. 将反应物倾入100 mL 冰水中, 调节pH 值至2, 析出大量红棕色针状晶体, 滤出晶体, 水洗, 干燥后得红棕色针状晶体11.5 g, 收率为76%, m.p. 143~144 ℃(文献值[8]: 143~144 ℃).1.3 2-羟基-4-甲氧基苯乙酮(5)的合成将2,4-二羟基苯乙酮(4) (1.520 g, 10 mmol)溶于无水丙酮溶液中, 在搅拌下加入无水K 2CO 3 (2.8 g, 20 mmol), 加热回流40 min 后降至40 ℃, 缓慢滴入硫酸二甲酯(1.4 mL, 15 mmol), 继续回流反应, TLC 检测反应. 将产物中丙酮蒸除, 然后用乙酸乙酯萃取, 用MgSO 4干燥, 硅胶柱分离[洗脱剂: V (石油醚)∶V (乙酸乙酯)=16∶1], 得淡黄色液体 1.38 g, 产率83%, m.p. 49~50 ℃(文献值[9] 50 ℃). 1.4 2,4-二羟基苯甲醛(6)的合成将间苯二酚(11 g, 0.1 mol)和7.3 g 二甲基甲酰胺(DMF)加入100 mL 三颈瓶中, 机械搅拌下, 逐渐加入7.6 mL POCl 3加完后, 继续搅拌2 h 得稠状液体. 反应混合物用热的75 mL 50% NaOAc 液溶解. 待冷却后, 产物用乙醚提取, 醚层用水洗, 无水MgSO 4干燥. 醚层减压蒸馏(沸点220 ℃), 蒸干后产物用水重结晶、烘干得乳白色针状晶体10.63 g, 产率77%, m.p. 134~136 (℃文献值[10] 134~136 )℃.1.5 2-甲氧甲氧基-4-甲氧基苯甲醛(7)的合成取910 mg 3 (6.6 mmol)溶于20 mL 丙酮中, 剧烈搅拌下加入 3.63 g 无水K 2CO 3, 0.7 mL (7.4 mmol) (CH 3)2SO 4, 回流5 h 后停止反应, 乙酸乙酯萃取, 水和饱和食盐水分别洗涤, 无水MgSO 4干燥, 硅胶柱层析分离[洗脱剂: V (石油醚)∶V (乙酸乙酯)=16∶1], 得白色固体810 mg, 产率81%, m.p. 39~40 ℃(文献值[11] 40~41 ℃); 1H NMR (CDCl 3, 400 MHz) δ: 3.86 (s, 3H, OCH 3), 6.53 (d, J =8.6 Hz, 1H, C 6-H), 6.41 (s, 1H, C 3-H), 7.38 (d, J =8.6 Hz, 1H, C 5-H), 9.72 (s, 1H, CHO), 11.50 (s, 1H, C 2-OH); IR (KBr) ν: 3160, 2967, 1659, 1632, 1221, 1113, 1022, 986, 842, 801 cm -1.将2-羟基-4-甲氧基苯甲醛(380 mg, 2.5 mmol)溶于2 mL CH 2Cl 2中, 加入含NaOH 200 mg (4.0 mmol)的水溶液和93 mg 碘化四丁基铵, 搅拌20 min 后, 滴入0.22 mL (3 mmol) MOMCl, 继续搅拌0.5 h 停止反应, 乙酸乙酯萃取, 水和饱和食盐水分别洗涤, 无水MgSO 4干燥, 蒸除溶剂, 硅胶柱层析分离[洗脱剂: V (石油醚)∶920有机化学V ol. 28, 2008V(乙酸乙酯)=8∶1], 得固体物质480 mg, 产率98%, m.p. 52~54 ℃(文献值[11]: 54~56 ℃); 1H NMR (CDCl3, 400 MHz) δ: 3.48 (s, 3H, OCH3), 3.61 (s, 3H, OCH3), 5.24 (s, 2H, OCH2O), 6.56 (d, J=8.6 Hz, 2H, ArH), 6.49~6.68 (m, 3H, ArH), 7.77 (d, J=8.6 Hz, 2H, ArH), 10.28 (s, 1H, CHO); IR (KBr) ν: 2964, 1665,1598, 1152, 1098, 1071 cm-1.1.6 2'-羟基-4,4'-甲氧基-2-二甲氧甲氧基查尔酮(8)的合成将酮5 (996 mg, 6 mmol)与醛8 (980 mg, 5 mmol)溶于乙醇5 mL中, 冰水冷却至0 ℃, 搅拌下缓慢滴加0℃的含有(KOH, 1.95 g)的水/乙醇4 mL溶液, 0 ℃反应1 h, 升至室温继续搅拌反应35 h, 将反应液倒入冰水中,用3 mol•L-1的盐酸调pH值约为2~3. 用乙酸乙酯提取(30 mL×3), 有机层合并, 依次用水、饱和食盐水洗涤, MgSO4干燥, 蒸除溶剂, 硅胶柱层析分离[洗脱剂: V(石油醚)∶V(乙酸乙酯)=8∶1], 得粘稠状液体 1.40 g, 产率81%. TCL检测反应.1H NMR (CDCl3, 400 MHz) δ: 3.37 (s, 1H, OH), 8.21 (d, J=15.6 Hz, 1H, Hβ), 7.83 (d, J=8.8 Hz, 1H, ArH), 7.63 (d, J=8.4, 1H, ArH), 7.57 (d, J=15.6 Hz, 1H, Hα), 6.75 (d, J=2.8 Hz, 1H, ArH), 6.61 (d, J=8.8 Hz, 1H, ArH), 6.48 (d, J=8.4 Hz, 1H, ArH), 6.49 (d, J=2.4 Hz, 1H, ArH), 5.29 (s, 2H, OCH2O), 3.84 (s, 6H, OCH3), 3.46 (s, 3H, OCH3); IR(KBr) ν: 3441, 1606, 1216 cm-1; MS (70 eV) m/z (%):344 ([M]+, 1.0), 299 (2.0), 282 (3.5), 222 (1.7), 196 (2.7), 177 (3.1), 161 (14), 151 (14.3), 45 (100).1.7 2'-羟基-4,4'-二甲氧基-2-二甲氧甲氧基二氢查尔酮(9)的合成将制得的查尔酮8 (832 mg, 2.4 mmol)溶于乙酸乙酯中, 加入催化的Pd-C (5%, 41 mg). 室温下搅拌加氢, TLC检测反应, 滤除催化剂(硅胶短柱), 蒸除溶剂, 残余物用硅胶柱分离[洗脱剂: V(石油醚)∶V(乙酸乙酯)=8∶1], 得无色液体678 mg, 产率81%. 1H NMR(CDCl3, 400 MHz) δ: 12.88 (s, 1H, OH), 7.68 (d, J=8.0 Hz, 1H, ArH), 7.09 (d, J=8.0 Hz, 1H, ArH), 6.70 (d, J=2.4 Hz, 1H, ArH), 6.49 (d, J=8.4 Hz, 1H, ArH), 6.40 (d, J=2.0 Hz, 1H, ArH), 5.20 (s, 2H, OCH2O), 3.84 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.48 (s, 3H, OCH3), 3.17 (t, J=8.0 Hz, 2H, CH2), 2.98 (t, J=8.0 Hz, 2H, CH2); IR(KBr) ν: 3442, 1608, 1213 cm-1; MS (70 eV) m/z (%):346 ([M]+, 2.3), 284 (2.1), 196 (0.93), 178 (3.6), 168 (4.2), 151 (52.4), 136 (15.7), 137 (14.7), 45 (100). 1.8 1,3-二-(2-羟基-4-甲氧基苯基)丙烷(1)的合成将2 mg Hg溶于1 mL水中, 与100 mg锌粉共同搅拌30 min, 倾去水溶液, 沉淀用水漂洗, 得到锌汞齐. 向配有搅拌器及回流冷凝器的三口瓶中, 加上述锌-汞齐、水(1 mL)及HCl (1 mL)混匀, 加入二氢查尔酮9 (100 mg, 0.29 mmol)和乙醇溶液(5 mL)搅拌, 加热, 回流至反应完全, TLC检测反应. 用乙酸乙酯提取(30 mL×3), 有机层合并, 依次用水、饱和食盐水洗涤, MgSO4干燥, 蒸除溶剂, 硅胶柱层析分离[洗脱剂: V(石油醚)∶V(乙酸乙酯)=4∶1], 得到白色固体68 mg, 产率74%, m.p. 68~72 ℃(文献值[7] 68~72 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.02 (d, J=8.0 Hz, 4H, H-6',6"), 6.45 (dd, J=8.0, 2.8 Hz, 2H, H-5',5"), 6.37 (d, J=2.4 Hz, 2H, H-3',3"), 3.75 (s, 6H, OCH3), 2.59 (t, J=7.6 Hz, 4H, H-1,3), 1.89 (t, J=8.0 Hz, 2H, H-2); IR (KBr) ν: 3410, 2933, 1616, 1515, 1283, 831 cm-1; MS (70 eV) m/z (%): 288 ([M]+, 5), 164 (4), 151 (12), 137 (35), 43 (100).1.9 1,3-二-(2,4-二羟基苯基)丙烷(2)的合成在氮气保护下向化合物1 (26 mg, 0.09 mmol)加入二氯甲烷2 mL, 充入搅拌溶解, 滴加含有0.5 mmol的BBr3二氯甲烷溶液1 mL, 冰水浴搅拌2 h. 反应完别后, 将反应液倒入冰水中, 二氯甲烷萃取, 硅胶柱层析分离[洗脱剂: V(石油醚)∶V(乙酸乙酯)=2∶1], 得白色固体20 mg, 产率85%, m.p. 177~180 ℃(文献值[7]: 179~181 ℃); 1H NMR (400 MHz, CD3OD) δ: 6.83 (d, J=7.6 Hz, 2H, H-3',3"), 6.25 (d, J=2.4 Hz, 2H, H-6', 6"), 6.19 (dd, J=8.2, 2.4 Hz, 2H, H-5',5"), 2.49 (t, J=7.6 Hz, 4H, H-1,3), 1.64 (t, J=7.6 Hz, 2H, H-2); IR (KBr) ν: 3354, 2922, 1616, 1516, 1219, 841 cm-1; MS (70 eV) m/z (%): 260 ([M]+, 14), 181 (4), 137 (37), 123 (100).2 结果与讨论2,4-二羟基苯乙酮按文献[8]方法由间苯二酚合成, 收率为76%, 按文献[9]方法将2,4-二羟基苯乙酮和硫酸二甲酯在丙酮中, 在无水碳酸钾作用下, 得到2-羟基-4-甲氧基苯乙酮(5), 产率83%; 2,4-二羟基苯甲醛按文献[10]方法由间苯二酚合成, 产率77%; 在无水碳酸钾作用下2,4-二羟基苯甲醛与硫酸二甲酯反应, 得到2-羟基-4-甲氧基苯甲醛, 产率81%, 在相转移催化剂碘化四丁基铵和氢氧化钠作用下, 2-羟基-4-甲氧基苯甲醛与氯甲基醚反应, 得到4-甲氧基-2-甲氧甲氧基苯甲醛(7), 产率98%; 由化合物5与化合物7羟醛缩合按文献[12]的方法, 在氢氧化钾的水/乙醇(V∶V=1∶1)溶液作用下, 反应36 h, 以81%的产率得到查耳酮8[12].No. 5 杨金会等:天然产物1,3-二-(2-羟基-4-甲氧基苯基)丙烷和1,3-二-(2,4-二羟基苯基)丙烷的合成921查耳酮8在乙酸乙酯溶液中, 在Pd-C催化下加氢还原, 反应3 h, 得到双氢查尔酮9, 产率81%, 这是合成的关键步骤, 需要注意的是该反应产物与反应时间和温度有关, 反应时间过短, 原料反应不完全, 反应时间过长, 有副反应发生, 副产物为产物羰基进一步还原为羟基得到的化合物. 经过多次实验发现, 在0 ℃下, 在乙酸乙酯中反应3 h, 效果最好.双氢查尔酮9用锌-汞齐还原, 将羰基还原为亚甲基, 同时脱除甲氧甲氧(MOMO)保护基, 得到目标化合物1, 需要指出的是此步反应温度不宜过高, 文献报道锌-汞齐还原羰基通常在加热条件下进行, 但是, 在加热条件下进行, 没有得到目标化合物, 经过多次试验发现, 在室温, 不加热条件下还原可以顺利得到目标化合物1, 产率74%, 原因可能是反应体系酸性很强, 在加热条件下, 化合物可能脱去了酚羟基上的甲基. 化合物1在二氯甲烷中, 在BBr3作用下, 脱除甲基得到目标化合物2, 产率85%. 化合物1和2波谱数据与文献[7]报道一致.完成了1,3-二-(2-羟基-4-甲氧基苯基)丙烷(1)和1,3-二-(2,4-二羟基苯基)丙烷(2)两个天然产物的首次全合成, 提供了一条简便的合成二芳基丙烷的方法.References1 Morais, A. A.; Braz-Filho, R.; Gottlieb, O. R. Phytochemistry1985, 24, 3023.2 Almeida, P. A.; Fraiz, S. V. Jr.; Braz-Filho, R. J. Braz.Chem. Soc. 1999, 10(5), 347.3 Braz-Filho, R.; Leite, M. F. F.; Gottlieb, O. R. Phytochemi-stry1973, 12(2), 417.4 Braz-Filho, R.; De Diaz, P. P.; Gottlieb, O. R. Phytochemi-stry1980, 19, 455.5 Braz-Filho, R.; Gottlieb, O. R.; Pinho, S. L. V. Phytochemi-stry1976, 15, 567.6 Takasugi, M.; Kumagai, Y.; Nagao, S. Chem. Lett. 1980,1459.7 Yang, S. L.; Liu, X. K. Chin. Chem. Lett. 2005, 16, 57.8 Liao, T. G.; Wang, Q. A.; Fang, W. Q.; Zhu, H. J. Chin. J.Org. Chem. 2006, 26, 685 (in Chinese).(廖头根, 汪秋安, 方伟琴, 朱华结, 有机化学, 2006, 26, 685.)9 Yu, M.-X.; Shao, Z.-L.; Dai, M.; Zhang, X.-F. Anhui Chem.Ind.1998, 24(5), 22 (in Chinese).(俞明兴, 邵宗龙, 戴玫, 张雪峰, 安徽化工, 1998, 24(5),22.)10 Mendelson, W. L.; Hayden, S. Synth. Commun. 1996, 26(3),603.11 Shao, H. W.; Wei, H. X.; Li, Y. L. Chin. J. Synth. Chem.1995, 3(4), 314 (in Chinese).(邵华武, 韦汉勋, 李裕林, 合成化学, 1995, 3(4), 234.) 12 Yang, Y. G.; Zhang, Y.; Cao, X. P. Acta Chim. Sinica2005,63(20), 1901 (in Chinese).(杨永刚, 张宇, 曹小平, 化学学报, 2005, 63(20), 1901.)(Y0709224 QIN, X. Q.; DONG, H. Z.)。

天然产物全合成学院:化学化工学院系别:化学系姓名:***学号:**************简介:天然产物全合成是有机化学中最为活跃、最具原动力的研究方向之一。
这方面的研究极大地推动了有机新反应、新方法、新试剂、新理论和新概念的发现和发展。
天然产物全合成也是发现、发展新医药等功能物质的重要途径,在医药健康、生命、材料以及能源等科学领域具有广阔的应用前景。
天然产物全合成是以天然产物(源自植物、动物或微生物的有机化合物)为目标分子,通过设计研究合成策略、路线和方法,从简单原料出发实现其化学合成。
研究内容主要包括:(1)高效、简捷和高选择性合成策略;(2)不对称(特别是催化不对称)合成策略;(3)选择廉价、易得的天然产物为原料,研究简捷、高效的半合成策略;(4)目标分子生物活性、结构多样化导向的合成策略;(5)针对目标分子关键结构(或骨架)的合成方法学研究,实现其形式合成;(6)生物催化和仿生合成。
关键词:天然产物、全合成、前言:天然产物全合成是一项难度大、耗资多、周期长、见效慢的工作,需要科学家集全面而深厚的有机化学知识、坚忍不拔的耐力和良好的综合素质于一身。
只要投入足够的财力和资源,建立客观合理的评价体系,就会有越来越多的学者投身到这项事业,中国的天然产物全合成研究就有可能走在世界的前列,并推动有机化学学科及相关产业的快速发展。
天然产物全合成是有机化学中最为活跃、最具原动力的研究方向之一。
这方面的研究极大地推动了有机新反应、新方法、新试剂、新理论和新概念的发现和发展,并在很大程度上体现了有机化学学科的发展水平和实力。
因此,一方面,天然产物全合成在有机化学的发展中仍将发挥无可替代的作用,具有更加辉煌的发展前景;另一方面,天然产物全合成也是发现和发展新医药等功能物质的重要途径,其所建立的方法同样也适用于其他有机物的制备,例如有机光电磁材料、高分子单体、组装体基元、有机探针分子、染料敏化剂。
因此,天然产物的化学合成研究在医药健康、生命、材料、能源等科学领域具有广阔的应用前景。


中国科学技术大学 博士学位论文 (1)天然产物CassiarinsA和B的全合成(2)喜树碱类药物SN38及 其新衍生物的合成 姓名:姚元山 申请学位级别:博士 专业:有机化学 指导教师:姚祝军 2011-04-20摘要摘要(1)疟疾是一种很古老的疾病,其对人类的危害已有数千年的历史。
即使 是文明高度发达的现代社会每年仍有数百万的人死于这种以蚊虫叮咬作为主要 传染途径的疾病。
在过去的几个世纪里,人类在抗疟斗争中取得了辉煌成就但并 没有根除这一恶疾。
与此相反,近年来发现疟原虫对已有的抗疟药物交叉抗药性 越来越严重,因此发展新的抗疟药物重新被赋予了重要的意义。
2007 年,日本科学家 Morita 等从印尼豆属植物铁刀木(Cassia siamea)的 叶中分离到两个结构新颖、具有优良抗疟活性并且低毒性的生物碱 cassiarins A 和 B。
根据它们的结构特点,我们从同一起始原料出发,成功地发展了三条不同 的全合成路线,其中包括一条仿生全合成路线。
在此基础上,我们也完成了三个 cassiarin A 衍生物的合成。
(2)目前,癌症正越来越严重地威胁人类的生命健康。
由其导致的死亡人 数也在逐年增加。
随着现代药物和生物化学的发展,化学药物在癌症治疗中占据 了越来越重要的地位。
喜树碱是由 Wall 等人在 1966 年从喜树中分离得到的具有优良抗癌活性的五 环生物碱。
虽然喜树碱的临床研究并不成功,但是作为先导化合物它为我们的抗 癌药物研究指明了方向。
目前,喜树碱家族在售的抗癌药物有伊立替康 (Irinotecan)和拓扑替康(Topotecan) ,并且有许多化合物处于临床研究的不同 阶段。
考虑到喜树碱药物的工业化生产对天然资源的依赖性以及发展新的喜树碱 衍生物药物的需要,我们小组于 2007 年和 2008 年先后发展了两条温和、高效的 喜树碱全合成路线。
本论文在此基础上实现了伊立替康活性代谢物 SN38 的全合 成和 30 个新喜树碱衍生物的合成。
天然产物的化学合成返回知识介绍首页在过去的十年里,天然产物的多级全合成始终向复杂分子的新水平上推进。
目前化学家们正在应付有机化学的挑战:即在镜像体中如何选择合成所需要的构象。
也就是选择合成特殊的手性中心。
化学家们正在重新确定研究的前沿目标和开辟一个新的有效进攻对象。
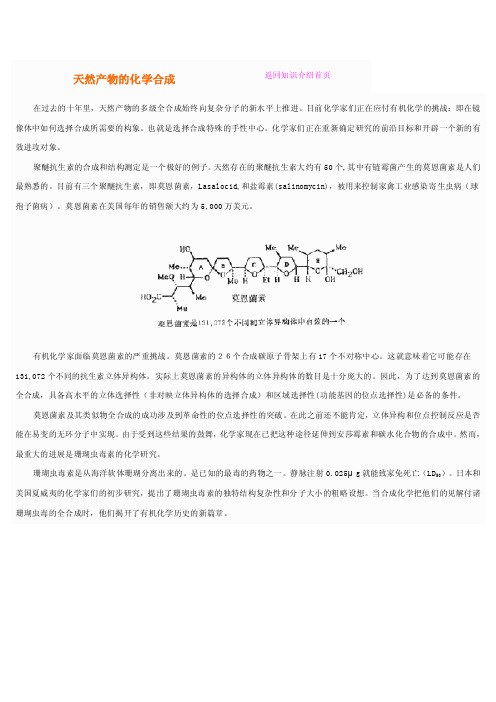
聚醚抗生素的合成和结构测定是一个极好的例子。
天然存在的聚醚抗生素大约有50个,其中有链霉菌产生的莫恩菌素是人们最熟悉的。
目前有三个聚醚抗生素,即莫恩菌素,Lasalocid,和盐霉素(salinomycin),被用来控制家禽工业感染寄生虫病(球孢子菌病)。
莫恩菌素在美国每年的销售额大约为5,000万美元。
有机化学家面临莫恩菌素的严重挑战。
莫恩菌素的26个合成碳原子骨架上有17个不对称中心。
这就意味着它可能存在131,072个不同的抗生素立体异构体。
实际上莫恩菌素的异构体的立体异构体的数目是十分庞大的。
因此,为了达到莫恩菌素的全合成,具备高水平的立体选择性(非对映立体异构体的选择合成)和区域选择性(功能基因的位点选择性)是必备的条件。
莫恩菌素及其类似物全合成的成功涉及到革命性的位点选择性的突破。
在此之前还不能肯定,立体异构和位点控制反应是否能在易变的无环分子中实现。
由于受到这些结果的鼓舞,化学家现在已把这种途径延伸到安莎霉素和碳水化合物的合成中。
然而,最重大的进展是珊瑚虫毒素的化学研究。
珊瑚虫毒素是从海洋软体珊瑚分离出来的。
是已知的最毒的药物之一。
静脉注射0.025μg就能致家免死亡(LD50)。
日本和美国夏威夷的化学家们的初步研究,提出了珊瑚虫毒素的独特结构复杂性和分子大小的粗略设想。
当合成化学把他们的见解付诸珊瑚虫毒的全合成时,他们揭开了有机化学历史的新篇章。
珊瑚虫毒素这个奇特的大分子有128个碳原子,其中64个处于不对称中心。
这些不对称中心由7个双链骨架连接起来,产生2×1021个立体构体。
由此看来,建立珊瑚虫毒素的立体化学是通向全合成的第一步。

天然产物的全合成策略研究论文素材天然产物作为药物研发的重要来源之一,具有广泛的生物活性和药理学价值。
然而,由于其天然来源的限制和结构的复杂性,在实际应用中经常面临供应不足和高成本的问题。
因此,针对天然产物的全合成策略研究具有重要的理论与应用意义。
1. 天然产物和全合成研究的背景天然产物是生物多样性的体现,分为植物、动物和微生物来源的天然产物。
具有广谱抗癌、抗菌、抗炎症、免疫调节等多种药理活性。
然而,由于天然产物获得途径的限制,扩大化学库对复杂结构化合物的全合成具有重要意义。
2. 天然产物全合成的意义与挑战天然产物的全合成具有以下意义:- 能够得到天然产物及其结构类似物的结构优化,以提高其活性和选择性。
- 增加药物的生物利用度,降低副作用。
- 提供足够的化合物样本,以供临床前研究和药物筛选使用。
然而,天然产物的全合成也面临以下挑战:- 复杂的碳骨架和多功能基团的引入。
- 低收率和高成本的问题。
- 需要耗费大量的时间和精力。
3. 天然产物全合成的策略和技术为了解决天然产物全合成的挑战,研究人员提出了一系列策略和技术:- 反应选择性的提高与合成路径的优化。
- 进一步了解天然产物的合成途径和生物活性。
- 开发新的合成方法和技术,如金属催化反应、手性催化剂等。
- 运用计算化学和机器学习等方法,辅助合成路径设计。
4. 天然产物全合成的成功案例通过不断的研究和探索,已经取得了一些天然产物全合成的成功案例,如:- Artemisinin(青蒿素)的全合成。
- Taxol(紫杉醇)的全合成。
- Corynanthean桥环类化合物的全合成。
这些成功案例为天然产物全合成提供了宝贵的经验和参考。
5. 结论天然产物的全合成策略研究对于药物研发和应用具有重要意义。
通过合理设计的全合成路径和优化的合成方法,可以解决天然产物供应不足和高成本的问题,为药物研发提供更多的选择和机会。

天然产物化学的全合成研究天然产物一直是药物研究领域的热点,因为许多药物都是从天然产物中提取出来的。
但是,由于天然产物结构复杂、含有多个手性中心,基本上无法通过化学合成来得到足够纯度和足够数量的产物。
因此,全合成研究成为天然产物化学的一个重要分支。
全合成研究的目标是通过合成尽可能类似于天然产物的化合物,以便研究其生物活性和药用价值。
但这并不是简单将天然产物的结构公式从头开始合成,而是从天然产物结构的某个部分开始,不断逼近目标化合物。
因此,全合成研究的成功不仅需要高超的化学技术,还需要洞察整个天然产物的结构和功能。
全合成研究的成功需要解决多个困难。
首先,需要找到一种经济高效的合成途径。
其次,由于天然产物大多具有手性中心,因此合成过程中需要控制手性,以便得到具有良好生物活性的具有手性的化合物。
此外,不同的场景下,需要合成出不同的产物,因此需要设计多种不同的全合成路线。
在过去的几十年中,许多成功的全合成研究案例已经在天然产物领域中形成。
例如,纳塔鲁克西环素全合成的实现,为学习其在细胞生长控制机制中的作用,提供了独特机会。
同时,香豆素全合成,为针对癌症和HIV治疗提供了新途径。
除此之外,阿霉素的全合成,为人们研究其抗生素活性、肝炎和艾滋病的治疗提供了很多新方法。
在全合成研究中,还有一种重要的策略叫做“生物或酶催化的合成”。
这种策略是利用天然产物的生物合成机制来合成越来越完整的模拟天然产物的分子。
在这个过程中,酶是执行化学反应的主要催化剂。
这种策略已经在苯丙素生物合成、初级胺生物合成和萜类化合物合成的全合成研究中被证明是很有前途的。
总的来说,全合成研究已经极大地推动了天然产物领域的发展。
在今后的研究中,科学家们还将面临更加复杂的问题和挑战,但随着技术的进步和研究经验的积累,相信会有更多更有价值的研究成果出现。

JACS:达特茅斯学院Micalizio课题组完成Anhydroryanodol的全合成天然产物全合成为有机化学的发展提供了平台,为设计和开发新化学反应提供了灵感。
Ryanodine及其水解产物ryanodol是从南美植物Ryana speciosa Vahl分离得到的高度官能团化的二萜类化合物(Figure 1),可以调节Ca2+离子通道。
1967年,Wiesner报道了ryanodine的结构是由五个不同的环组成,包含11个手性中心,其中8个是连续全取代的sp3碳原子。
最早,Deslongchamps课题组报道了ryanodol的合成,其关键步骤涉及Diels-Alder反应、Baeyer-Villiger氧化和新颖的氧化裂解/跨环aldol级联反应。
上述研究激发了合成化学家们对立体电子效应在有机化学中发挥作用的兴趣。
Inoue 课题组利用自由基烯丙基化、钯催化的烯烃异构化和闭环易位化学等策略完成了ryanoid的合成。
最近,Reisman课题组通过铑催化的Pauson-Khand反应和SeO2介导的多步氧化过程等关键转化实现了ryanodol的步骤经济性合成。
近日,美国达特茅斯学院Glenn C. Micalizio课题组开发了一种概念独特的碳环构建方法,用于(+/–)-anhydroryanodol的合成,其为ryanodine的降解产物,也是ryanodol合成中常用的后期中间体(Figure 1),该成果发表于近期J. Am. Chem. Soc.(DOI: 10.1021/jacs.0c05766)。
(图片来源:J. Am. Chem. Soc.)Ryanodol(1)的合成策略(Figure 2):ryanodol可以追溯至处于高度氧化态的四环化合物2,其可以由三环二环氧化物3通过以下过程获得:1)缩醛转化为内酯;2)皂化、C11位选择性环氧开环;3)通过闭环烯烃复分解构建C环。

谈天然药物化学史话:奎宁的发现、化学结构以及全合成本文从网络收集而来,上传到平台为了帮到更多的人,如果您需要使用本文档,请点击下载按钮下载本文档(有偿下载),另外祝您生活愉快,工作顺利,万事如意!奎宁(quinine)是非常著名的天然药物,曾经挽救了无数人的生命,甚至被认为影响了人类的发展进程和天然产物全合成进程的重大发现。
对奎宁的研究在科学史上也留下了非常重要的记录,20 世纪,有4 位科学家因在与疟疾相关的研究中做出杰出贡献而获得了诺贝尔化学奖以及生理学或医学奖。
奎宁的发现过程非常偶然和有趣,其立体结构的确定曾被认为是结构鉴定的一个经典范例,尤其是奎宁的全合成被认为是开创了立体选择性反应(stereoselective reaction)的先河。
在继重要天然药物紫杉醇、银杏内酯、岩沙海葵毒素、河豚毒素的总结之后,本文对奎宁的发现、结构鉴定、生物活性和全合成进行简要介绍,以纪念在奎宁的研究中做出伟大贡献的科学家,同时为科研人员在复杂天然产物全合成工作中开阔眼界、拓宽思路提供一些帮助。
1 奎宁的发现奎宁俗称金鸡纳碱,属于来自天然的生物碱类(alkaloids)化合物,最早是从茜草科植物金鸡纳树Cinchona ledgeriana (Howard) Moens ex 及其同属植物的树皮中提取得到的。
奎宁是治疗疟疾的特效药,它的发现及应用曾经挽救了无数疟疾病人的生命。
奎宁的真实起源目前并无实证,但是民间印第安人用金鸡纳树皮泡水来治疗发热高烧,也就是现在的疟疾。
约四百多年前欧洲殖民者侵略美洲时,很多欧洲人不适应当地的气候条件,染上了严重的疟疾而死亡。
当时,西班牙驻秘鲁总督的夫人安娜(Ana Chinchón)也不幸染上了疟疾,这时一位印第安姑娘冒着生命危险给安娜夫人偷偷送去了金鸡纳树皮制成的粉末,安娜夫人服用后,转危为安。
后来一位西班牙传教士将金鸡纳树皮带到了西班牙,并将树皮取名为cincnona。


生物天然产物的全合成研究自然界中存在大量的生物天然产物,它们广泛存在于各种植物、动物和微生物中,具有丰富的结构类型和生物活性。
然而,这些天然产物通常只以微量量产,无法满足广泛的研究需求和医药应用。
因此,人们通过全合成的方法来制备这些天然产物,以满足科学研究和药物开发的需要。
全合成是指利用有机合成化学方法,从简单的化合物出发,逐步构建天然产物的复杂结构。
全合成的成功与否,除了需要合适的化学方法外,还需要足够的知识和技能。
特别是对于天然产物的结构、性质和生物活性的全面理解,对全合成的实现至关重要。
全合成不仅可以提供足量的天然产物用于各种应用,还可以制备结构类似但活性更强、更稳定的合成产物,以期望能发现全新的天然产物和药物。
全合成中的一项重要工作是合成中间体,它们是全合成化学反应的关键步骤,决定了后续的结构构建和转化。
例如,合成多环天然产物时,常常需要制备具有多个手性中心和不同官能团的复杂分子。
因此,中间体的构建对于天然产物全合成至关重要。
许多人通过化学合成的途径来合成这些复杂中间体,其中一些途径涉及大量的化学步骤,且导致产率不高。
因此,有很多研究人员正在探索合成更有效和经济的中间体的方法。
另外,生物合成方法也被广泛用于合成天然产物全合成过程中。
这些方法利用生物合成中已存在的酶促反应来构建目标天然产物的结构,这些酶都是生物体内已存在的天然催化剂。
相比于传统的化学合成,生物合成具有多种优点,例如反应选择性高、副反应少、反应条件温和、对环境的影响较小。
因此,生物合成在全合成中也扮演着重要的角色。
总之,天然产物的全合成是有机合成领域中的重大挑战之一。
通过利用不同的合成策略,包括化学合成、生物合成等多种途径,我们可以合成出具有复杂结构和生物活性的药物分子。
全合成的科学研究不断取得突破性进展,在未来的研究中,我们可以期望通过全合成来制备出更有效、更安全的药物,以满足人们对健康的更强需求。