中国药品检验标准操作规范2010年版熔点测定
- 格式:doc
- 大小:84.50 KB
- 文档页数:11

XXXXXXXXX 有限公司质量控制管理制度1目的:本规程规范了熔点仪的自校验方法,确保熔点仪使用的检验结果的有效性及准确性,保证检验结果的准确可靠。
2范围:本规程适用于质保部熔点仪的自校准。
3职责:质保部QC人员4引用标准:《中国药典》2015年版,《中国药品检验标准操作规范》2010年版。
5标准物质:校准用的标准品有标准物质奈(终熔806C)、标准物质己二酸(终熔1529C)、标准物质蒽醌(终熔2857C)。
6可接受标准:测量示值误差:小于200C为士05C; 200〜300C为士08C。
7校准程序7.1首先对仪器管管座等位置进行清洁,擦拭,保证仪器不受污染物影响,达到最佳使用状态。
然后开启电源开关。
7.2将传温液杯放入适量传温液。
7.3接通电源,按下电源开关,仪器即处于复位状态,在复位状态,按〖+〗、〖-〗两键设置数码管显示的预置温度为低于待测标准品熔点约10C,升温速率为1.0C /min,按下“预置”键,仪器即处于准备状态,当传温液到达预置温度,处于稳定WRF型熔点仪自校准操作规程第2页共2页的温度状态后,可放入标准品。
8 样品准备8.1将标准品置于瓷研钵内,轻轻研碎成尽可能细密的粉末,以得到均一样品。
8.2每个标准品用3支清洁、干燥的熔点管,将其开口端插入标准品种,分别装入样品。
8.3取一长约0.8m的干燥玻璃管,直立于玻璃板上,将装有标准品的熔点管在其中投落至少20次,使熔点管内样品紧缩至3mm高。
9校准方法9.1将装有样品的毛细管插在样品架上,放入传温液。
9.2观察毛细管中样品熔化过程,分别按下〖初熔1〗、〖终熔1〗、〖初熔2〗、〖终熔2〗、〖初熔3〗、〖终熔3〗键记录两个熔点值。
9.3样品测试后,按下“预置”键,传温液即开始降温,至温度预置值,进行下一次测试。
9.4每一标准品重复测定三次,连续的3根标准品终熔值很集中,即重复性好。
10自校准周期熔点仪的自校准周期为一年,仪器维修之后可增加自校准次数。

红外分光光度法1 简述化合物受红外辐射照射后,使分子的振动和转动运动由较低能级向较高能级跃迁,从而导致对特定频率红外辐射的选择性吸收,形成特征性很强的红外吸收光谱,红外光谱又称振-转光谱。
红外光谱是鉴别物质和分析物质化学结构的有效手段,已被广泛应用于物质的定性鉴别、物相分析和定量测定,并用于研究分子间和分子内部的相互作用。
习惯上,往往把红外区分为3个区域,即近红外区(12800~4000cm,0.78~2.5μm)。
其中中红外区是药物分析中最常用的区域。
红外吸收与物质浓度的关系在一定范围内服从于朗伯-比尔定律,因而它也是红外分光光度法定量的基础。
红外分光光度计分为色散型和傅里叶变换型两种。
前者主要由光源、单色器(通常为光栅)、样品室、检测器、记录仪、控制和数据处理系统组成。
以光栅为色散元件的红外分光光度计,以波数为线性刻度,以棱镜为色散元件的仪器,以波长为线性刻度。
波数与波长的换算关系如下:波数(cm-1)= 104波长(μm)傅里叶变换型红外光谱仪(简称FT-IR)则由光学台(包括光源、干涉仪、样品室和检测器)、记录装置和处理系统组成,由干涉图变为红外光谱需经快速傅里叶变换。
该型仪器现已成为最常用的仪器。
2 红外分光光度计的检定所用仪器应按现行国家质量与核查技术监督局“色散型红外分光光度计检定规程”、“傅里叶变换红外光谱仪检定规程”和《中国药典》附录规定,并参考仪器说明书,对仪器定期进行校正检定。
2.1 波数准确度2.1.1波数准确度的允差范围傅里叶变换红外光谱仪在3000cm-1附近的波数误差应不大于±5cm-1,在1000cm-1附近的波数误差应不大于±1cm-1。
2.1.2波数准确度检定方法2.1.2.1以聚苯乙烯膜校正按仪器使用说明书要求设置参数,以常用的扫描速度记录厚度为50μm的聚苯乙烯膜红外光谱图。
测量有关谱带的位置,其吸收光谱图应符合《药品红外光谱集》所附聚苯乙烯图谱的要求,并与参考波数(表1)比较,计算波数准确度。
目的:建立熔点测定法标准操作规程。
适用范围:熔点测定。
责任:质检员实施本操作规程,检验室主任负责监督本规程实施。
程序:1.简述熔点系指一种物质按照规定的方法测定由固相熔化成液相时的温度,是物质的一项物理常数。
依法测定熔点,可以鉴别或检查药品的纯杂程度。
根据被测物质的不同性质,在中国药典2000年版二部附录Ⅵ C“熔点测定法”项下列有三种不同的测定方法,分别用于测定易粉碎的固体药品、不易粉碎的固体药品、或凡士林及其类似物质,并在正文各该品种项下明确规定应选用的方法;遇有在正文未注明方法时,均系指采用第一法。
在第一法中,又因熔融时是否同时伴有分解现象,现时规定有不同的升温速度和观测方法。
由于因测定方法,受热条件和判断标准的不同,常导致测得的结果有明显的差异,因此在测定时,必须根据药典正文各该品种项下的规定选用方法,并严格遵照该方法中规定的操作条件和判断标准进行测定,才能获得准确的结果。
本规程仅适用于中国药典2000年版二部附录Ⅵ C“熔点测定法”中的第一法与第二法,而不适用于第三法。
2.仪器与用具2.1加热用容器硬质高型玻璃烧杯,或可放入内热加热器的大内径园底玻璃管,供盛装传温液用。
2.2搅拌器电磁搅拌器,或用垂直搅拌的杯状玻璃搅拌棒,用于搅拌加热的传温液,使之温度均匀。
2.3温度计具有0.5℃刻度的分浸型温度计,其分浸线的高度宜在50mm至80mm之间(分浸线低于50mm的,因汞球距离液面太近,易受外界气温的影响;分浸线高于80mm 的,则毛细管容易漂浮;均不宜使用),温度计的汞球宜短,汞球的直径宜与温度计柱身的粗细接近(便于毛细管装有供试品的部位能紧贴在温度计汞球上)。
温度计除应符合国家质量技术监督局的规定外,还应经常采用药品检验用“熔点标准品”进行校正。
2.4毛细管系用洁净的中性硬质玻璃管拉制而成,内径为0.9~1.1mm,壁厚为0.10~0.15mm,分割成长10cm以上;最好将两端熔封,临用时再锯开其一端(用于第一法)或两端(用于第二法),以保证毛细管内洁净干燥。

1.目的建立熔点检验操作规程,保证检验人员操作规范化、标准化,确保本公司的产品质量2.适用范围适用于本公司原料达卡巴嗪、甘露醇等熔点测定。
3.责任本文件由QA负责起草,质量部部长审核,质量管理负责人批准。
QC检验人员负责本规程的实施。
4.引用标准《中国药典》2010年版二部5.内容5.1.仪器与用具熔点测定仪毛细管表面皿玻璃管干燥器称量瓶烧杯5.1.1.温度计:温度计除应符合国家质量技术监督局的规定外,还应经常采用药品检验用“熔点标准品”进行校正。
(本公司定为每3个月对使用点进行一次校准)5.2.操作原理在常温常压下,物质受热时,从固态转变成液态,固态和液态达到平衡状态相互共存时的温度,就是该物质的熔点。
5.3.传温液与熔点标准品5.3.1.传温液5.3.1.1.水:用于测定熔点在80℃以下者。
用前应先加热至沸使脱气,并放冷。
5.3.1.2.硅油:熔点介于80~200℃之间者,用黏度不小于50mm2/s的硅油;熔点高于200℃者,用黏度不小于100mm2/s的硅油。
5.3.2.药品检验用熔点标准品:由中国药品生物制品检定所供应,专供测定熔点时校正温度计用(见表1)。
用前应在研钵中研细,并按所附说明书中规定的条件干燥后,置五氧化二磷干燥器中避光保存备用。
表1 熔点标准品5.4.操作方法5.4.1.取供试品,研成细粉,除另有规定外,按照其项下干燥失重的条件进行干燥。
若该供试品为不检查干燥失重,熔点范围低限在135℃以上,受热不分解的供试品可采用105℃干燥;熔点在135℃或受热分解的供试品可在五氧化二磷干燥器中干燥。
干燥过夜或用其他事宜的干燥方法干燥,如恒温减压干燥。
分取供试品规定项下量,置熔点测定用毛细管(简称毛细管,由中性硬质玻璃管制成,长9cm以上,内径0.9~1.1mm,壁厚0.10~0.15mm,一端熔封;当所用温度计浸入传温液在6cm 以上时,管长应适当增加, 使露出液面3cm以上)中,轻击管壁或借助长短适宜的洁净玻璃管,垂直放在表面皿上,将毛细管自上口放入使自由落下,反复数次,使粉末紧密集结在毛细管的熔封端。

气相色谱-质谱联用法1 简述气相色谱-质谱联用法(GC-MS)将高效的气相色谱技术与能够提供丰富结构信息和专属性定量结果的质谱技术相结合,广泛应用于易挥发的或经衍生化处理后易挥发的有机物分析。
GC-MS法语LC-MS法互补,已成为药物研究、生产、临床检测的重要技术手段。
2 仪器组成及原理GC-MS联用仪由图1所示的各部分组成。
图1 气相色谱-质谱联用仪组成框图气相色谱仪在大气压下分离待测样品中的各组分;接口把气相色谱流出的各组分导入处于真空状态的质谱仪,起着气相色谱和质谱之间适配器的作用;质谱作为气相色谱的检测器,将分离后的各组分分别离子化、质量分析、离子检测;计算机系统用于气相色谱、接口和质谱仪的控制,同时进行数据采集和处理。
2.1 进样方式常采用直接进样或色谱分离后进样方式。
2.1.1 直接进样微量注射器将少量的待测化合物溶液经接口导入质谱仪分析。
2.1.2 分离后进样经气相色谱分离后的不同组分,部分或全部经接口导入质量仪分析。
2.2 接口GC-MS接口是解决气相色谱和质谱联用的关键组建。
质谱离子源的真空度在10-3Pa,而GC色谱柱出口压力高达105Pa,接口的作用就是要使两者压力匹配。
理想的接口是既能除去全部载气,又能把待测化合物从气相色谱仪传导质谱仪。
直接导入型接口(interface of direct coupling)灵敏度高,传输率100%,广泛应用于毛细管气相色谱-质谱联用。
其工作原理示意如图2,待测组分与载气(氦气)一起从内径为0.25~0.32mm的毛细管气相色谱柱内流出,不发生电离,被真空泵抽走,而待测组分被电离、形成各种离子,进一步质谱分析。
接口的实际作用是支撑插入端毛细管,使其准确定位,以及保持温度,使色谱柱流出物不发生冷凝。
具有低流速的毛细管气相色谱柱很容易与现代质谱仪相匹配1~2ml/min的速度。
2.3 离子源气相色谱-质谱联用仪中最常用的离子化方法为电子轰击离子化(Electron Ionization,EI)和的化学离子化(Chemical Ionization,CI)。

铅、镉、砷、汞、铜测定法---原子吸收分光光度法1 简述本法系采用原子吸收分光光度法对中药材中的铅、镉、砷、汞、铜进行限量检查。
2 仪器与用具2.1 原子吸收分光光度计应配备有火焰原子化器、石墨炉原子化器和适宜的氢化物发生装置,并具有氘灯或塞曼效应背景校正功能;铅、镉、砷、汞、铜等元素的空心阴极灯;普通或热解涂层石墨管;乙炔气、高纯氩气或高纯氮气;空气压缩机及冷却循环水泵等。
2.2 微波消解仪内罐为聚四氟乙烯材料制成,具有适宜的耐压密封装置和过压安全保护装置;具有程序控制、功率可调的微波发生装置;可采用适宜的方式监控反应罐内的温度和压力。
2.3 电热板应具有温度均匀的加热表面和温度控制装置。
2.4 纳氏比色管或量瓶应尽可能使用耐腐蚀的塑料器具,以聚四氟乙烯材料制成的为好,玻璃器皿易吸附或吸收金属离子,因此仅适于短时间内对溶液的容量使用。
3 试药与试液3.1 铅、镉、砷、汞、铜单元素标准溶液及国家一级标准物质杨树叶中国剂量科学研究院提供,单元素标准溶液用于制备标准曲线,杨树叶或茶树叶可作为工作对照物质,检查方法的可靠性。
3.2 硝酸、高氯酸应采用高纯试剂,盐酸、硫酸、磷酸二氢铵、硝酸镁为优级纯,碘化钾、抗坏血酸、盐酸羟胺为分析纯,使用前应检查各试剂中的相关金属元素含量符合测定的要求。
3.3 水去离子水或用石英蒸馏器蒸馏的超纯水,使用前应检查其中的相关金属元素含量符合测定的要求。
3.4 25%碘化钾溶液取碘化钾25g,加水100ml使溶解,即得。
本液应临用新制。
3.5 10%抗坏血酸溶液取抗坏血酸10g,加水100ml使溶解,即得。
本液应临用新制。
3.6 含1%磷酸二氢铵溶液和0.2%硝酸镁溶液的混合溶液取磷酸二氢铵1g,硝酸镁0.2g,加水100ml使溶解,即得。
3.7 1%硼氢化钠和0.3%氢氧化钠混合溶液取氢氧化钠3g,加水1000ml使溶解,加入硼氢化钠3g,使溶解,即得。
本液应临用新制。
3.8 4%硫酸溶液取硫酸4ml,加入水中稀释,并加水至100ml,即得。
药品检验结果允差范围评价准则
1、目的
检验结果的质量是实验室工作的重点,为保证检验结果的准确、科学和有效,应对检验结果进行有效评价,检验过程中会出现系统误差和随机误差,误差影响着检验结果的准确性和精密度。
为了判定检验结果有效、可信,特制订药品检验主要检测项目允差范围。
2 、范围
适用本所的检验数据结果的判定。
3 、依据
《中国药品检验操作规范》2010年版、《中国药典》2010年版
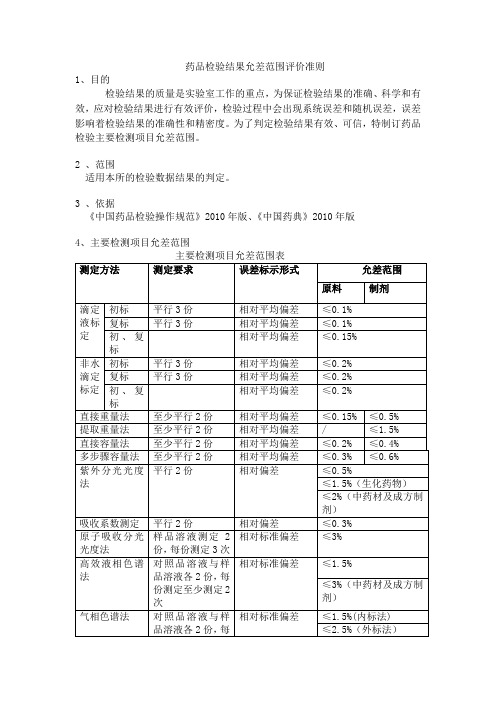
4、主要检测项目允差范围
本所进行的质量控制,以及平时检验工作中应遵循药品检验结果允差的评价准则。


熔点测定法中国药品检验标准操作规范XXXX版熔点测定方法1简介熔点是指物质按照规定的方法从固相熔化成液相的温度,是物质的物理常数。
依法测定熔点可以鉴别或检查药物的纯度。
根据被测物质的不同性质,中国药典XXXX版二部附录ⅵC“熔点测定法”中有三种不同的测定方法,分别用于测定易粉碎的固体药物、不易粉碎的固体药物、凡士林及类似物质,并明确规定了每类下的选择方法。
如果品种下没有规定方法,则采用第一种方法。
在第一种方法中,由于熔化是否伴随着分解,规定了不同的加热速率和观察方法。
由于测量方法、加热条件和判断标准的不同,测量结果往往有明显的差异。
因此,在测定时,必须根据药典中各品种的规定选择方法,并严格遵守方法中规定的操作条件和判断标准,以获得准确的结果。
仪器和用具2.1加热容器是一个坚硬的高型玻璃烧杯,或者可以放入一个内部加热加热器的大内径圆底玻璃管中,用于保存温度转移液体。
2.2搅拌器电磁搅拌器或立式搅拌器环形玻璃搅拌器用于搅拌加热的温度转移液体,使其温度均匀。
2.3温度计用0.5℃刻度的浸没式温度计,浸没线高度应在50毫米至80毫米之间(浸没线小于50毫米,因为水银球太靠近液面,易受外界空气温度的影响;如果浸润线高于80毫米,毛细管将很容易浮动。
没有人适当使用),温度计的水银球应短,水银球的直径应接近温度计柱体的厚度(便于毛细管装有测试条的部分可接近温度计的水银球)。
温度计应符合国家质量技术监督局的规定,并定期用药品检验的“熔点标准”进行校正。
2.4毛细管从干净的中性硬玻璃管中拉出,其内径为0.9-1.1毫米,壁厚为0.10-0.15毫米,分割长度为9厘米或更长,毛细管的一端熔合(对于第一种方法)或毛细管的一端未熔合(对于第二种方法);当使用的温度计浸入6厘米以上的温度转移液体中时,应适当增加管子长度,以暴露3厘米以上的液位。
两端也可以熔合和密封,毛细管的一端(对于第一种方法)或两端(对于第二种方法)在使用时可以锯掉,以确保毛细管内部的清洁和干燥。

1.主题内容:建立有熔点测定法操作方法。
2.适用范围:本规程适用于检查药物在生产过程中的熔点测定法的操作。
3.引用标准:《中国药典2010版一部》
4.责任:化验员、QC主管。
5. 用途:化验室
6.内容:依照待测物质的性质不同,测定法分为下列3种。
各品种项下未注明时,均系指第一法。
6.1第一法:测定易粉碎的固体药品
取供试品适量,研成细粉,除另有规定外,应按照各品种项下干燥失重的条件进行干燥。
若该品种为不检查干燥失重、熔点范围低限在135℃以上、受热不分解的供试品,可在五氧化二磷干燥器中干燥过夜或用其他适宜的干燥方法干燥,如恒温减压干燥。
分取供试品适量,置熔点测定用毛细管(简称毛细管,由中性硬质玻璃管制成,长9cm以上,内径0.9~1.1mm,壁厚0.10~0.15mm,一端熔封;当所有温度计浸入传温液在6cm以上时,管长应适当增加,使露出液面3cm以上)。

中国药品检验标准操作规范2010年版滴定液1.0 简述1. 1 滴定液系指在容量分析中用于滴定被测物质含量的标准溶液,具有准确的浓度(取4 位有效数字)。
1. 2 滴定液的浓度以“mol/L”表示,其基本单元应符合药典规定。
1. 3 滴定液的浓度值与其名义值之比,称为“_F”值,常用于容量分析中的计算。
1 . 4 本操作规范适用于《中国药典》20 1 0年版二部附录X V F“滴定液”的配制与标定。
2 .0仪器与用具2 . 1 分析天平其分度值(感量)应为O . l m g或小于O.lmg;毫克组砝码需经校正,并列有校正表备用。
2.2 10、2 5和5 0 m l滴定管应附有该滴定管的校正曲线或校正值。
2.3 10、15、2 0和2 5 m l移液管其真实容量应经校准,并附有校正值。
2.4 2 5 0 m l和1 0 0 0 m l量瓶应符合国家A 级标准,或附有校正值。
3.0 试药与试液3 . 1 均应按照《中国药典》附录X V F“滴定液”项下的规定取用。
3 . 2 基准试剂应有专人负责保管与领用。
4.0 配制滴定液的配制方法有间接配制法与直接配制法两种,应根据规定选用,并应遵循下列有关规定。
4.1所用溶剂“水”,系指蒸馏水或去离子水,在未注明有其他要求时,应符合《中国药典》“纯化水”项下的规定。
4.2采用间接配制法时,溶质与溶剂的取用量均应根据规定量进行称取或量取,并且制成后滴定液的浓度值应为其名义值的0 . 9 5〜1.05;如在标定中发现其浓度值超出其名义值的0 . 9 5〜1.05范围时,应加人适量的溶质或溶剂予以调整。
当配制量大于1000ml时,其溶质与溶剂的取用量均应按比例增加。
4.3采用直接配制法时,其溶质应采用“基准试剂”,并按规定条件干燥至恒重后称取,取用量应为精密称定(精确至4 〜5 位有效数字),并置1000ml量瓶中,加溶剂溶解并稀释至刻度,摇匀。
配制过程中应有核对人,并在记录中签名以示负责。

1.目的建立熔点检验操作规程,保证检验人员操作规范化、标准化,确保本公司的产品质量2.适用范围适用于本公司原料达卡巴嗪、甘露醇等熔点测定。
3.责任本文件由QA负责起草,质量部部长审核,质量管理负责人批准。
QC检验人员负责本规程的实施。
4.引用标准《中国药典》2010年版二部5.内容5.1.仪器与用具熔点测定仪毛细管表面皿玻璃管干燥器称量瓶烧杯5.1.1.温度计:温度计除应符合国家质量技术监督局的规定外,还应经常采用药品检验用“熔点标准品”进行校正。
(本公司定为每3个月对使用点进行一次校准)5.2.操作原理在常温常压下,物质受热时,从固态转变成液态,固态和液态达到平衡状态相互共存时的温度,就是该物质的熔点。
5.3.传温液与熔点标准品5.3.1.传温液5.3.1.1.水:用于测定熔点在80℃以下者。
用前应先加热至沸使脱气,并放冷。
5.3.1.2.硅油:熔点介于80~200℃之间者,用黏度不小于50mm2/s的硅油;熔点高于200℃者,用黏度不小于100mm2/s的硅油。
5.3.2.药品检验用熔点标准品:由中国药品生物制品检定所供应,专供测定熔点时校正温度计用(见表1)。
用前应在研钵中研细,并按所附说明书中规定的条件干燥后,置五氧化二磷干燥器中避光保存备用。
表1 熔点标准品5.4.操作方法5.4.1.取供试品,研成细粉,除另有规定外,按照其项下干燥失重的条件进行干燥。
若该供试品为不检查干燥失重,熔点范围低限在135℃以上,受热不分解的供试品可采用105℃干燥;熔点在135℃或受热分解的供试品可在五氧化二磷干燥器中干燥。
干燥过夜或用其他事宜的干燥方法干燥,如恒温减压干燥。
分取供试品规定项下量,置熔点测定用毛细管(简称毛细管,由中性硬质玻璃管制成,长9cm以上,内径0.9~1.1mm,壁厚0.10~0.15mm,一端熔封;当所用温度计浸入传温液在6cm 以上时,管长应适当增加, 使露出液面3cm以上)中,轻击管壁或借助长短适宜的洁净玻璃管,垂直放在表面皿上,将毛细管自上口放入使自由落下,反复数次,使粉末紧密集结在毛细管的熔封端。

2010版中国药典凡例总则一、《中华人民共和国药典》简称《中国药典》,依据《中华人民共和国药品管理法》组织制定和颁布实施。
《中国药典》一经颁布实施,其同品种的上版标准或其原国家标准即同时停止使用。
《中国药典》由一部、二部、三部及其增补本组成,内容分别包括凡例、正文和附录。
除特别注明版次外,《中国药典》均指现行版《中国药典》。
本部为《中国药典》二部。
二、国家药品标准由凡例与正文及其引用的附录共同构成。
本部药典收载的凡例、附录对药典以外的其他中药国家标准具同等效力。
三、凡例是为正确使用《中国药典》进行药品质量检定的基本原则,是对《中国药典》正文、附录及与质量检定有关的共性问题的统一规定。
四、凡例和附录中采用的“除另有规定外”这一用语,表示存在与凡例或附录有关规定不一致的情况时,则在正文中另作规定,并按此规定执行。
五、正文中引用的药品系指本版药典收载的品种,其质量应符合相应的规定。
六、正文所设各项规定是针对符合《药品生产质量管理规范》(Good Manufacturing Practices, GMP)的产品而言。
任何违反GMP 或有未经批准添加物质所生产的药品,即使符合《中国药典》或按照《中国药典》没有检出其添加物质或相关杂质,亦不能认为其符合规定。
七、《中国药典》的英文名称为Pharmacopoeia of The People’s Republic of China, 英文简称Chinese Pharmacopoeia;英文缩写为Ch.P.。
正文八、正文系根据药物自身的理化与生物学特性,按照批准的处方来源、生产工艺、贮藏运输条件等所制定的、用以检测药品质量是否达到用药要求并衡量其质量是否稳定均一的技术规定。
九、正文项下根据品种和剂型不同,按顺序可分别列有:(1)品名(包括中文名称、汉语拼音与英文名);(2)有机药物的结构式;(3)分子式与分子量;(4)来源或有机药物的化学名称;(5)含量或效价规定;(6)处方;(7)制法;(8)性状;(9)鉴别;(10)检查;(11)含量或效价测定;(12)类别;(13)规格;(14)贮藏;(15)制剂等。

熔点测定法标准操作规程1 简述熔点系指一种物质按照规定的方法测定由固相熔化成液相时的温度,是物质的一项物理常数。
依法测定熔点,可以鉴别或检查药品的纯杂程度。
根据被测物质的不同性质,在《中国药典》2010年版二部附录Ⅵ C“熔点测定法”项下列有三种不同的测定方法,分别用于测定易粉碎的固体药品、不易粉碎的固体药品、或凡士林及其类似物质,并在正文各该品种项下明确规定应选用的方法;遇有在正文未注明方法时,均系指采用第一法。
在第一法中,又因熔融时是否同时伴有分解现象,而规定有不同的升温速度和观测方法。
由于测定方法,受热条件和判断标准的不同,常导致测得的结果有明显的差异,因此在测定时,必须根据药典正文各该品种项下的规定选用方法,并严格遵照该方法中规定的操作条件和判断标准进行测定,才能获得准确的结果。
2 仪器与用具2.1 加热用容器硬质高型玻璃烧杯,或可放入内热式加热器的大内径园底玻璃管,供盛装传温液用。
2.2 搅拌器电磁搅拌器,或用垂直搅拌的杯状玻璃搅拌棒,用于搅拌加热的传温液,使之温度均匀。
2.3 温度计具有0.5℃刻度的分浸型温度计,其分浸线的高度宜在50mm至80mm之间(分浸线低于50mm 的,因汞球距离液面太近,易受外界气温的影响;分浸线高于80mm的,则毛细管容易漂浮;均不宜使用),温度计的汞球宜短,汞球的直径宜与温度计柱身的粗细接近(便于毛细管装有供试品的部位能紧贴在温度计汞球上)。
温度计除应符合国家质量技术监督局的规定外,还应经常采用药品检验用“熔点标准品”进行校正。
2.4 毛细管系用洁净的中性硬质玻璃管拉制而成,内径为0.9~1.1mm,壁厚为0.10~0.15mm,分割成长9cm以上;一端熔封(用于第一法)或管端不熔封(用于第二法);当所用温度计浸入传温液在6cm以上时,管长应适当增加,使露出液面3cm以上。
也可将两端熔封,临用时再锯开其一端(用于第一法)或两端(用于第二法),以保证毛细管内洁净干燥。

文件内容:1、主题内容和适用范围 (2)2、引用标准 (2)3、简介 (2)4、第一法光阻法 (2)5、第二法显微计数法 (5)6、更改信息 (7)颁发部门:质量管理部。
分发清单:QC办公室、中药室、化学室、稳定性考察室。
1 主题内容和适用范围本程序规定了不溶性微粒的检查方法和注意事项,使其规范化、标准化,并描述了更改信息。
本程序适用于检查溶液型静脉用注射液中不溶性微粒的大小和数量。
2 引用标准中国药典2010年版二部附录Ⅸ C“不溶性微粒检查法”、中国药品检验标准操作规“不溶性微粒检查法”。
范2010年版P2493 简介测定方法分两种即光阻法和显微计数法。
一般先采用光阻法;当光阻法测定结果不符合规定或供试品不适用于光阻法测定时,应采用显微计数法进行测定,并以显微计数法的测定结果为最终判定依据。
4 第一法光阻法4.1实验环境、仪器与用具实验环境实验操作所处环境应不得导入明显的微粒,测定前的操作在层流净化台中进行。
玻璃仪器和其他所需的用品都应洁净,无微粒。
本法所用微粒检查用水(或其他适宜溶剂),使用前须经不大于1.0µm的微孔滤膜滤过。
仪器装置光阻法不溶性微粒测定仪通常包括定量取样器、传感器和数据处理器三部分。
测量粒度范围为2~100µm,检测微粒浓度为0~10000个/ml。
测定仪应定期校正与检定(至少每6个月校正一次),并符合规定。
4.2操作程序4.2.1检查前准备使用适宜的清洁仪器,取50ml微粒检查用水(或其他溶剂)经微孔滤膜(一般孔径为0.45µm)滤过,置于洁净的适宜容器中,旋转使可能存在的微粒均匀,静置待气泡消失。
按光阻法项下的检查法检查,每10ml中含10µm以上(≥10um)的不溶性微粒应在10粒以下,含25µm以上(≥25um)的不溶性微粒应在2粒以下。
否则表明微粒检查用水(或其他溶剂)、玻璃仪器和实验环境不适于进行微粒检查,应重新进行处理,检测符合规定后方可进行供试品检查。

中药片剂片剂系指提取物、提取物加饮片细粉或饮片细粉与适宜辅料混匀压制或用其他适宜方法制成的圆片状或异形片状的制剂,有浸膏片、半浸膏片和全粉片等。
片剂以口服普通片为主,另有含片、咀嚼片、泡腾片、阴道片、阴道泡腾片和肠溶片等。
对片剂的质量要求,除外观应完整光洁,色泽均匀,有适宜的硬度,以及各品种项下规定的检查项目外,还应检查“重量差异”、“崩解时限”和“微生物限度”。
此外,阴道片还应检查“融变时限”,阴道泡腾片检查“发泡量”。
“重量差异”检查法1 简述本法适用于片剂的重量差异检查。
2 仪器与用具2.1 分析天平感量0.1mg(适用于平均片重在0.30g以下的片剂)或感量1mg(适用于平均片重0.30g或0.30g以上的片剂)。
2.2 扁形称量瓶。
2.3 弯头或平头手术镊。
3 操作方法3.1 取空称量瓶,精密称定重量;再取供试品20片,置此称量瓶中,精密称定。
两次称量值之差即为20片供试品的总重量,除以20,得平均片重(m)。
3.2 从已称定总重量的20片供试品中,依次用镊子取出1片,分别精密称定重量,得各片重量。
4 注意事项4.1 在称量前后,均应仔细查对药片数。
称量过程中,应避免用手直接接触供试品。
已取出的药片,不得再放回供试品原包装容器内。
4.2 遇有检出超出重量差异限度的药片,宜另器保存,供必要时的复核用。
4.3 糖衣片应在包衣前检查片芯的重量差异,符合规定后方可包衣。
包衣后不再检查重量差异。
4.4 薄膜衣片在包衣后也应检查重量差异。
5 记录与计算5.1 记录每次称量数据。
5.2 求出平均片重(m),保留三位有效数字。
5.3 按下表规定的重量差异限度,求出允许片重范围(m±m×重量差异限度)。
平均重量重量差异限度0.3g以下±7.5%0.3g或0.3g以上±5%5.4 遇有超出允许片重范围并处于边缘者,应再与标示片量或平均片量相比较,计算出该片重量差异的百分率,再根据上表规定的重量差异限度作为判定的依据(避免在计算允许重量范围时受数值修约的影响)。

熔点测定法标准操作规程目的:建立熔点测定法标准操作规程。
适用范围:熔点测定。
责任:质检员实施本操作规程,检验室主任负责监督本规程实施。
程序:1.简述熔点系指一种物质按照规定的方法测定由固相熔化成液相时的温度,是物质的一项物理常数。
依法测定熔点,可以鉴别或检查药品的纯杂程度。
根据被测物质的不同性质,在中国药典2000年版二部附录Ⅵ C“熔点测定法”项下列有三种不同的测定方法,分别用于测定易粉碎的固体药品、不易粉碎的固体药品、或凡士林及其类似物质,并在正文各该品种项下明确规定应选用的方法;遇有在正文未注明方法时,均系指采用第一法。
在第一法中,又因熔融时是否同时伴有分解现象,而规定有不同的升温速度和观测方法。
由于测定方法,受热条件和判断标准的不同,常导致测得的结果有明显的差异,因此在测定时,必须根据药典正文各该品种项下的规定选用方法,并严格遵照该方法中规定的操作条件和判断标准进行测定,才能获得准确的结果。
本规程仅适用于中国药典2000年版二部附录Ⅵ C“熔点测定法”中的第一法与第二法,而不适用于第三法。
2.仪器与用具2.1加热用容器硬质高型玻璃烧杯,或可放入内热式加热器的大内径园底玻璃管,供盛装传温液用。
2.2搅拌器电磁搅拌器,或用垂直搅拌的杯状玻璃搅拌棒,用于搅拌加热的传温液,使之温度均匀。
2.3温度计具有0.5℃刻度的分浸型温度计,其分浸线的高度宜在50mm至80mm之间(分浸线低于50mm 的,因汞球距离液面太近,易受外界气温的影响;分浸线高于80mm的,则毛细管容易漂浮;均不宜使用),温度计的汞球宜短,汞球的直径宜与温度计柱身的粗细接近(便于毛细管装有供试品的部位能紧贴在温度计汞球上)。
温度计除应符合国家质量技术监督局的规定外,还应经常采用药品检验用“熔点标准品”进行校正。
2.4毛细管系用洁净的中性硬质玻璃管拉制而成,内径为0.9~1.1mm,壁厚为0.10~0.15mm,分割成长10cm以上;最好将两端熔封,临用时再锯开其一端(用于第一法)或两端(用于第二法),以保证毛细管内洁净干燥。

1、紫外-可见分光光度法(P58~59):含量测定时供试品应称取2份,如为对照品比较法,对照品一般也应称取2份。
吸收系数检查也应称取供试品2份,平行操作,每份结果对平均值的偏差应在±0.5%以内。
作鉴别或检查可取样品1份。
吸收系数测定法样品应同时测定2份,同一台仪器测定的2份结果,对平均值的偏差应不超过±0.3%,否则应重新测定。
2、原子吸收分光光度法(P70):供试品要求制备2份样品溶液,各测定3次。
取平均值从标准曲线上求得相应的浓度。
测定的相对标准偏差(RSD)应不大于3%,石墨炉法可适当放宽。
样品测定离散性大时应多测几次,以增加读数的可靠性。
《明胶药用空心胶囊铬检测方法指导原则》铬测定:由于微量测定,结果的偏差会较大,一般两份样品的相对偏差≤10%,取平均值即可。
3、荧光分析法(P73):2份供试品测定结果,每份结果对平均值的偏差应在±1.5%以内,否则应重做。
4、火焰光度法(P75):定量分析采用第一法或第二法时,要求制备2份供试品溶液,各测定3次,测定的相对标准偏差(RSD)应不大于5%。
样品测定离散性大时应多测几次,以增加读数的可靠性。
5、高效液相色谱法(P81):供试品溶液与对照品溶液每份至少进样2次,由全部注样平均值(n ≥4)求得平均值,相对标准偏差(RSD)一般应不大于1.5%。
(此规定为05版药品操作规程上描述,10版无此规定)6、气相色谱法(P102):精密称取供试品和对照品各2份,按-----。
每份校正因子测定溶液(或对照品溶液)各进样2次,2份共4个校正因子相应值的平均标准偏差不得大于2.0%。
多份供试品测定时,每隔5批应再进对照品2次,供试品测定完毕,最后再进行对照品2次,核对下仪器有无改变。
7、毛细管电泳法(P111):标准品(对照品)溶液每份至少进样2次,由全部进样结果(n>4),求得平均值,相对标准偏差(RSD)一般应不大于3.0%。
文件内容:1、主题内容和适用范围 (2)2、引用标准 (2)3、简介 (2)4、仪器与用具 (2)5、传温液与熔点标准品 (3)6、操作程序 (4)7、结果与判定 (7)8、附注 (7)9、更改信息 (8)颁发部门:质量管理部。
分发清单:QC办公室、中药室、化学室、稳定性考察室。
1 主题内容和适用范围本程序规定了熔点的测定方法和影响因素,使其规范化、标准化,并描述了更改信息。
本程序适用于药品熔点的测定。
2 引用标准中国药典2010年版一部附录Ⅶ C和二部附录Ⅵ C “熔点测定法”、中国药品检验“熔点测定法”。
标准操作规范2010年版P1413 简介熔点系指一种物质按照规定的方法测定由固相熔化成液相时的温度,是物质的一项物理常数。
依法测定熔点,可以鉴别或检查药品的纯杂程度。
根据被测物质的不同性质,在中国药典2010年版附录Ⅵ C“熔点测定法”项下列有三种不同的测定方法,分别用于测定易粉碎的固体药品、不易粉碎的固体药品(如脂肪、脂肪酸、石蜡、羊毛脂等)、凡士林或其他类似物质,并在各该品种项下明确规定应选用的方法;遇有在品种项下未注明方法时,均系指采用第一法。
在第一法中,又因熔融时是否同时伴有分解现象,而规定有不同的升温速度和观测方法。
由于因测定方法、受热条件和判定标准的不同,常导致测得的结果有明显的差异,因此在测定时,必须根据各品种项下的规定选用方法,并严格遵照该操作程序中规定的操作条件和判定标准进行测定,才能获得准确的结果。
4 仪器与用具加热用容器硬质高型玻璃烧杯,或可放入内热式加热器的大内径圆低玻璃管,供盛装传温液用。
温度计具有0.5℃刻度的分浸型温度计,其分浸线的高度宜在50mm至80mm之间(分浸线低于50mm的,因汞球距离液面太近,易受外界气温的影响;分浸线高于80mm 的,则毛细管容易漂浮;均不易使用),温度计的汞球宜短,汞球的直径宜与温度计柱身的粗细接近(便于毛细管装有供试品的部位能紧贴在温度计汞球上)。
温度计除应符合国家质量技术监督局的规定外,还应经常采用药品检验用“熔点标准品”进行校正。
搅拌器电磁搅拌器,或用垂直搅拌的环状玻璃搅拌棒,用于搅拌加热的传温液,使之温度均匀。
毛细管系采用洁净的中性硬质玻璃管拉制而成,内径0.9~1.1mm,壁厚0.10~0.15mm,分割成长9cm以上,一端熔封(用于第一法)或管端不熔封(用于第二法);当所用温度计浸入传温液在6cm以上时,管长应适当增加,使露出液面3cm以上。
也可将两端熔封,临用时再锯开其一端(用于第一法)或两端(用于第二法),以保证毛细管内洁净干燥。
熔点测定仪带有电磁搅拌装置(用于搅拌加热的传温液,使之温度均匀),其温度探头应经常采用药品检验用“熔点标准品”进行校正。
5 传温液与熔点标准品5.1传温液水用于测定熔点在80℃以下者。
用前应先加热至沸使脱气,并放冷。
硅油熔点介于80~200℃之间者,用黏度不小于50mm2/s的硅油;熔点高于200℃者,用黏度不小于100mm2/s的硅油。
5.2药品检验用熔点标准品由中国药品生物制品检定所分发,专供测定熔点时校正温度计用。
用前应在研钵中研细,并按所附说明书中规定的条件干燥(见下表)后,置五氧化二磷干燥器中干燥避光保存备用。
熔点标准品注:上述熔点指全熔时的温度。
6 操作程序6.1第一法测定易粉碎的固体药品。
6.1.1供试品的预处理取供试品,置研钵中研细,移置扁形称量瓶中,按各品种项下“干燥失重”的条件进行干燥。
如该药品不检查干燥失重,则对熔点低限在135℃以上而受热不分解的品种,可采用105℃干燥;对熔点在135℃以下或受热分解的品种,可在无氧化二磷干燥器中干燥过夜。
个别品种在品种项下另有规定的,则按规定处理。
6.1.2将毛细管开口的一端插入上述预处理后的供试品中,再反转毛细管,并将熔封一端轻叩桌面,使供试品落入管底,再借助长短适宜(约60cm)的洁净玻璃管,垂直放在表面皿或其他适宜的硬质物体上,将上述装有供试品的毛细管放入玻璃管上口使其自由落下,反复数次,使供试品紧密集结于毛细管底部;装入供试品的高度应为3mm。
个别品种规定不能研磨、不能受热、并要减压熔封测定的,可将供试品少许置洁净的称量纸上,隔纸迅速用玻璃棒压碎成粉末,迅速装入毛细管使其高度达3mm;再将毛细管开口一端插入一根管壁有一小孔的耐压橡皮管的小孔中,橡皮管末端用玻璃棒密塞,另一端接在抽气泵上,在抽气减压的情况下熔封毛细管。
6.1.3将温度计垂直悬挂于加热用容器中,使温度计汞球的底端处于加热面(加热器)的上方2.5cm以上;加入适量的传温液,使传温液的液面约在温度计的分浸线处。
加热传温液并不断搅拌,俟温度上升至较规定的熔点低限尚低10℃时,调节升温速度使每分钟上升1.0~1.5℃(对于熔融时同时分解的供试品,则其升温速度为每分钟上升(2.5~3.0℃),待到达预计全熔的温度后降温;如此反复2~3次以掌握升温速度,并便于调整温度计的高度使其在全熔时的分浸线恰处于液面处。
6.1.4当传温液的温度上升至待测品种规定的熔点低限尚低10℃时,将装有供试品的毛细管浸入传温液使贴附(或用毛细管夹或橡皮圈固定)在温度计上,要求毛细管的内容物部分适在汞球的中部;根据6.1.3掌握升温速度,继续加热并搅拌,注意观察毛细管内供试品的变化情况;记录供试品在毛细管内开始局部液化并出现明显液滴时的温度作为初熔温度,全部液化时的温度作为全熔温度(全熔时毛细管内的液体应完全澄清。
个别药品在熔融成液体后会有小气泡停留在液体中,此时容易与未熔融的固体相混淆,应仔细辨别)。
凡在正文品种的熔点项下注明“熔融时同时分解”的品种,除升温速度应调节为每分钟上升2.5~3.0℃外,并应以供试品开始局部液化出现明显液滴或开始产生气泡时的温度作为初熔温度,以供试品的固相消失、全部液化时的温度作为全熔温度。
遇有固相消失不明显时,应以供试品分解物开始膨胀上升时的温度作为全熔温度;无法分辨初熔和全熔时,可记录其产生突变(例如颜色突然变深、供试品突然迅速膨胀上升)时的温度作为熔点。
此时可只有一个温度数据。
6.1.5影响因素传温液的升温速度,毛细管的内径和壁厚及其洁净与否,以及供试品装入毛细管内的高度及其紧密程度,均将影响测定结果,因此比须严格按照规定进行操作。
6.1.6测定过程中易出现的各种现象初熔之前,毛细管内的供试物可能出现“发毛”、“收缩”、“软化”、“出汗”等现象,在未出现局部液化的明显液滴和持续熔融过程时,均不作初熔判断。
但如上述现象严重,过程较长或因之影响初熔点的观察时,应视为供试品纯度不高的标志而予以记录;并身法与正常的该品种作对照测定,以便于最终判断。
“发毛”系指毛细管内的柱状供试物因受热而在其表面呈现毛糙;“收缩”系指柱状供试物子昂其中心聚集紧缩,或贴在某以边璧上;“软化”系指柱状供试物在收缩后变软,而形成软质柱状物,并向下弯塌;“出汗”系指柱状供试物收缩后在毛细管内壁出现细微液滴,但尚未出现局部液化的明显液滴和持续的熔融过程。
6.2第二法测定不易粉碎的固体药品(如脂肪、脂肪酸、石蜡、羊毛脂等)。
6.2.1取供试品,注意用尽可能低的温度使之熔融,另取两端锯开的毛细管,垂直插入上述熔融的供试品中,使供试品被吸入毛细管内的高度达10±1mm,取出后,擦去毛细管外壁的残留物,在10℃以下的冷处放置24小时,或置冰上放冷不少于2小时,使之完全凝固。
6.2.2将上述装有供试品的毛细管用橡皮圈固定在温度计上,使毛细管的内容物部分适在汞球的中部。
将毛细管连同温度计垂直浸入传温液(只能用水,液面距加热面应在6cm以上)中,并使供试品的上端适在传温液液面下10mm±1mm处(此时温度计的分浸线不可能恰在液面处,可不考虑)。
6.2.3缓缓加热并不断搅拌传温液,俟温度上升至较规定的熔点底线尚低5.0℃±0.5℃时,调节加温速率使每分钟升温0.3~0.5℃,注意观察毛细管内供试品的变化,检读供试品在毛细管内开始上升时的温度,即得。
6.3第三法测定凡士林或其他类似物质。
6.3.1供试品的预处理取供试品适量,缓缓搅拦并加热至温度达90~92℃,放入一平底耐热容器中使供试品的厚度为12mm±1mm,放冷至较规定的熔点上限高8~10℃.6.3.2用温度计黏附供试品事先取温度计插入试管所附的软木塞,并放冷至5℃,擦干。
待完成6.3.1的操作时,立即将放冷至5℃的温度计汞球部垂直插入经预处理的供试品中,直至碰到容器底部(即浸没12mm±1mm),随即取出温度计并保持垂直悬置,俟黏附在温度计汞球部的供试品表面浑浊,将温度计浸入16℃以下的水中5分钟,取出,将温度计插入一外径约25mm、长150mm试管中,塞紧固定软木塞于管口,使温度计悬于其中,并使温度计汞球部的底端距试管底部约15mm。
6.3.3近似熔点的测定将上述插入有温度计与供试品的试管垂直固定于水浴中,并使试管底与烧杯底的距离为10~20mm;然后在水浴内注入约16℃的水,至水浴液面与温度计的分浸线相平;加热水浴并缓缓搅拌,使水浴温度以每分钟上升2℃的速度升至38℃,再继续以每分钟上升1℃的速率升温至供试品的第一滴脱离温度计为止;立即检读温度计上显示的温度(估读至0.1℃),即为该供试品的近似熔点。
6.3.4测定结果取供试品,按6.3.1~6.3.3反复测定数次,如连续3次测得近似熔点的极差(最大值与最小值之差)未超过1.0℃时,即取3次的平均值(加上温度计的校正值)作为该供试品的熔点;如连续3次测得近似熔点的极差超过1.0℃时,可再测定2次,并取5次的平均值(加上温度计的校正值)作为该供试品的熔点。
7 结果与判定7.1对第一法中的初熔、全熔或分解突变时的温度,以及第二法中熔点的温度,都要估读到0.1℃,并记录突变时或不正常的现象。
每一检品应至少重复测定3次,3次读数的极差不大于0.5℃且不在合格与不合格边缘时,可取3次的均值加上温度计的校正值后作为熔点测定的结果。
如3次读数的极差为0.5℃以上时,或在合格与不合格边缘时,可在重复测定2次,并取五次的平均值加上温度计的校正值后作为熔点测定的结果。
必要时可选用正常的同一药品再次进行测定,记录其结果并进行比较。
7.2测定结果的数据应按修约间隔为0.5进行修约,即0.1~0.2℃舍去,0.3~0.7℃修约为0.5℃,0.8~0.9℃进为1℃;并以修约后的数据报告。
但当标准规定的熔点范围,其有效数字的定位为个位数时,则其测定结果的数据应修约间隔为1进行修约,即一次修约到标准规定的个位数。
7.3经修约后的初熔、全熔或分解突变时的温度均在各品种“熔点”项下规定的范围以内时,判为“符合规定”。
但如有下列情况之一者,即判为“不符合规定”:(1)初熔温度低于规定范围的低限;(2)全熔温度超过规定范围的高限;(3)分解点或熔点温度处于规定范围之外;(4)初熔前出现严重的“发毛”、“收缩”、“软化”、“出汗”现象,且其过程较长,并于正常的该药品作对照比较后有明显的差异者。