有机化学反应机理+范例+原理
- 格式:doc
- 大小:409.50 KB
- 文档页数:46
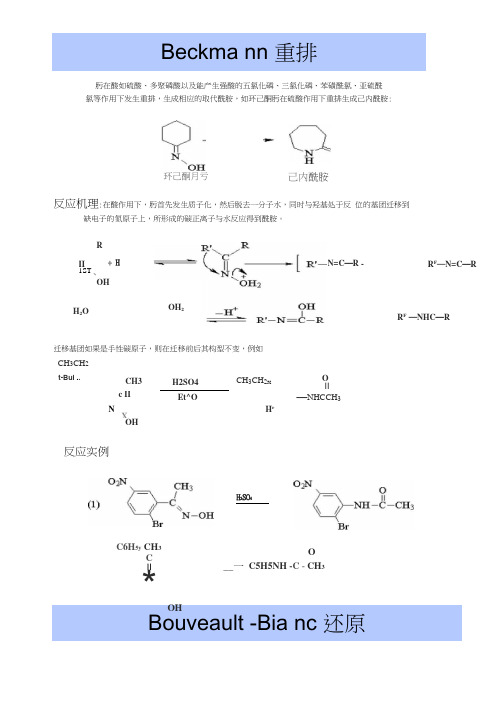
Beckma nn 重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰 氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:环己酮月亏己内酰胺Bouveault -Bia nc 还原反应机理:在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反 位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
R II 1ST、OH + H H 2O N=C —R -OH 2 R F —N=C —RR F —NHC —R迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如 CH 3CH 2t-Bui .. CH3 H2SO4 CH 3CH 2XOIIc IIEt^O—NHCCH 3N H rX OH反应实例 H 2SO 4C6H 5y CH 3CII*OHO__一 C5H5NH -C - CH 3脂肪族竣酸酯可用金属钠和醇圧原得一级醇。
氏不饱和竣酸酯 还原得相应的饱和醇°芳香酸酯也可进行本反应,但收率较低,本法 在氢化锂铝还原酯的方法发现以前,广泛地被使用,非共辄的双键可 不受影响9OEtOHR —C —OR 1 + 皿■ RCH 3OH + RQH反应机理首先酯从金属钠获得一个电子还原为自由基负离子,然后从醇中 夺取一个质子转变为自由基,再从钠得到一个电子生成负离子,泊除 烷细基成为醛,醛再经过相同的步骤还原咸醇钠,再酸化得到相应的 醇。
□O-EtCHR —C —OR' + Na ------------------------- — R^C —OR 1 _— R —C —OR 1_R —CH —OR* ------------ R —CH-OR ---------------------- R —C —H■ R —C —HiEtCHR —G —H ----------------Na +FCH 2OH反应实例醛酮也可以用本法还賦 得到相应的醇;Claisen- Schmidt 反应一个无:一氢原子的醛与一个带有:一氢原子的脂肪族醛或酮在稀氢氧化钠水溶液或醇 溶液存在下发生缩合反应,并失水得到:汀;不饱和醛或酮:反应机理NaR —CH 3(CHi )10CO^tNa EtOHCH/CH^KCHaOH 75%EtO 2C(CH^CO^t ——_■ EtOH HOCH XCH 抚CH 例畑CHgH^CHOC 哄⑴沖・叭如皿0HCH=CH-CHO 亠 H 2OClaise n 酯缩合反应含有僅-氢的酯在酚钠等碱性缩合剂作用下发生缩合作用,失去一分子 醇得到E 番同酸it 如2分子乙酸乙酯在金属钠和少量乙醇作用下发生缩合 得到乙麻乙酸乙酯。

一.Arbuzow反响(重排)亚磷酸三烷基酯作为亲核试剂与卤代烷感化,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反响时,其活性次序为:R'I >R'Br >R'Cl.除了卤代烷外,烯丙型或炔丙型卤化物.a-卤代醚.a- 或 b-卤代酸酯.对甲苯磺酸酯等也可以进行反响.当亚酸三烷基酯中三个烷基各不雷同时,老是先脱除含碳原子数起码的基团.本反响是由醇制备卤代烷的很好办法,因为亚磷酸三烷基酯可以由醇与三氯化磷反响制得:假如反响所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基雷同(即 R' = R),则 Arbuzow反响如下:这是制备烷基膦酸酯的经常运用办法.除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能产生该类反响,例如:反响机理一般以为是按 S N2 进行的分子内重排反响:反响实例二.Arndt-Eister 反响酰氯与重氮甲烷反响,然后在氧化银催化下与水共热得到酸.反响机理重氮甲烷与酰氯反响起首形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)产生重排得烯酮(3),(3)与水反响生成酸,若与醇或氨(胺)反响,则得酯或酰胺.反响实例三.Baeyer----Villiger 反响反响机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁徙到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时产生O-O键异裂.是以,这是一个重排反响具有光学活性的3---苯基丁酮和过酸反响,重排产品手性碳原子的枸型保持不变,解释反响属于分子内重排:不合错误称的酮氧化时,在重排步调中,两个基团均可迁徙,但是照样有必定的选择性,按迁徙才能其次序为:醛氧化的机理与此类似,但迁徙的是氢负离子,得到羧酸.反响实例酮类化合物用过酸如过氧乙酸.过氧苯甲酸.间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边拔出一个氧原子生成响应的酯,个中三氟过氧乙酸是最好的氧化剂.这类氧化剂的特色是反响速度快,反响温度一般在10~40℃之间,产率高.四.Beckmann 重排肟在酸如硫酸.多聚磷酸以及能产生强酸的五氯化磷.三氯化磷.苯磺酰氯.亚硫酰氯等感化下产生重排,生成响应的代替酰胺,如环己酮肟在硫酸感化下重排生成己内酰胺:反响机理在酸感化下,肟起首产生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁徙到缺电子的氮原子上,所形成的碳正离子与水反响得到酰胺.迁徙基团假如是手性碳原子,则在迁徙前后其构型不变,例如:反响实例五.Birch还原芬芳化合物用碱金属(钠.钾或锂)在液氨与醇(乙醇.异丙醇或仲丁醇)的混杂液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物.反响机理起首是钠和液氨感化生成溶剂化点子,然后苯得到一个电子生成自由基负离子(Ⅰ),这是苯环的л电子系统中有7个电子,加到苯环上谁人电子处在苯环分子轨道的反键轨道上,自由基负离子仍是个环状共轭系统,(Ⅰ)暗示的是部分共振式.(Ⅰ)不稳固而被质子化,随即从乙醇中牟取一个质子生成环己二烯自由基(Ⅱ).(Ⅱ)在取得一个溶剂化电子改变成环己二烯负离子(Ⅲ),(Ⅲ)是一个强碱,敏捷再从乙醇中牟取一个电子生成1,4-环己二烯.环己二烯负离子(Ⅲ)在共轭链的中央碳原子上质子化比末尾碳原子上质子快,原因尚不清晰.反响实例代替的苯也能产生还原,并且经由过程得到单一的还原产品.例如六.Bouveault---Blanc 还原脂肪族羧酸酯可用金属钠和醇还原得一级醇.α,β-不饱和羧酸酯还原得响应的饱和醇.芬芳酸酯也可进行本反响,但收率较低.本法在氢化锂铝还原酯的办法发明以前,广泛地被运用,非共轭的双键可不受影响.反响机理起首酯从金属钠获得一个电子还原为自由基负离子,然后从醇中牟取一个质子改变成自由基,再从钠得一个电子生成负离子,清除烷氧基成为醛,醛再经由雷同的步调还原成钠,再酸化得到响应的醇.反响实例醛酮也可以用本法还原,得到响应的醇:七.Bucherer 反响萘酚及其衍生物在亚硫酸或亚硫酸氢盐存鄙人和氨进行高温反响,可得萘胺衍生物,反响是可逆的.反响时如用一级胺或二级胺与萘酚反响则制得二级或三级萘胺.若有萘胺制萘酚,可将其参加到热的亚硫酸氢钠中,再参加碱,经煮沸除去氨而得.反响机理本反响的机理为加成清除进程,反响的第一步(无论从哪个偏向开端)都是亚硫酸氢钠加成到环的双键上得到烯醇(Ⅱ)或烯胺(Ⅵ),它们再进行下一步互变异构为酮(Ⅲ)或亚胺(Ⅳ):反响实例八.苯基羟胺(N-羟基苯胺)和稀硫酸一路加热产生重排成对-氨基苯酚:在H2SO4-C2H5OH(或CH3OH)中重排生成对-乙氧基(或甲氧基)苯胺:其他芳基羟胺,它的环上的o-p位上未被代替者会起类似的重排.例如,对-氯苯基羟胺重排成2-氨基-5-氯苯酚:反响机理反响实例九.Berthsen,A.Y 吖啶合成法二芳基胺类与羧酸在无水ZnCl2存鄙人加热起缩合感化,生成吖啶类化合物.反响机理反响机理不详反响实例十.Cannizzaro 反响凡α位碳原子上无生动氢的醛类和浓NaOH或KOH水或醇溶液感化时,不产生醇醛缩合或树脂化感化而起歧化反响生成与醛相当的酸(成盐)及醇的混杂物.此反响的特点是醛自身同时产生氧化及还原感化,一分子被氧化成酸的盐,另一分子被还原成醇:脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会产生此反响,其他醛类与强碱液,感化产生醇醛缩合或进一步变成树脂状物资.具有α-生动氢原子的醛和甲醛起首产生羟醛缩合反响,得到无α-生动氢原子的β-羟基醛,然后再与甲醛进行交叉Cannizzaro反响,如乙醛和甲醛反响得到季戊四醇:反响机理醛起首和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢负离子的情势转移到另一分子的羰基不克不及碳原子上.反响实例十一.Chichibabin 反响杂环碱类,与碱金属的氨基物一路加热时产生胺化反响,得到响应的氨基衍生物,如吡啶与氨基钠反响生成2-氨基啶,假如α位已被占领,则得γ-氨基吡啶,但产率很低.本法是杂环上引入氨基的轻便有用的办法,广泛实用于各类氮杂芳环,如苯并咪唑.异喹啉.丫啶和菲啶类化合物均能产生本反响.喹啉.吡嗪.嘧啶.噻唑类化合物较为艰苦.氨基化试剂除氨基钠.氨基钾外,还可以用代替的碱金属氨化物:反响机理反响机理还不是很清晰,可能是吡啶与氨基起首加成,(Ⅰ),(Ⅰ)转移一个负离子给质子赐与体(AH),产生一分子氢气和形成小量的2-氨基吡啶(Ⅱ),此小量的(Ⅱ)又可以作为质子的赐与体,最后的产品是2-氨基吡啶的钠盐,用水分化得到2-氨基吡啶:反响实例吡啶类化合物不轻易进行硝化,用硝基还原法制备氨基吡啶甚为艰苦.本反响是在杂环上引入氨基的轻便有用的办法,广泛实用于各类氮杂芳环,如苯并咪唑.异喹啉.吖啶和菲啶类化合物均能产生本反响.十二.Claisen 酯缩合反响含有α-氢的酯在醇钠等碱性缩合剂感化下产生缩合感化,掉去一分子醇得到β-酮酸酯.如2分子乙酸乙酯在金属钠和少量乙醇感化下产生缩合得到乙酰乙酸乙酯.二元羧酸酯的分子内酯缩合见Dieckmann缩合反响.反响机理乙酸乙酯的α-氢酸性很弱(pK a-24.5),而乙醇钠又是一个相对较弱的碱(乙醇的pK a~15.9),是以,乙酸乙酯与乙醇钠感化所形成的负离子在均衡系统是很少的.但因为最后产品乙酰乙酸乙酯是一个比较强的酸,能与乙醇钠感化形成稳固的负离子,从而使均衡朝产品偏向移动.所以,尽管反响系统中的乙酸乙酯负离子浓度很低,但一形成后,就不竭地反响,成果反响照样可以顺遂完成.经常运用的碱性缩合剂除乙醇钠外,还有叔丁醇钾.叔丁醇钠.氢化钾.氢化钠.三苯甲基钠.二异丙氨基锂(LDA)和Grignard试剂等.反响实例假如酯的α-碳上只有一个氢原子,因为酸性太弱,用乙醇钠难于形成负离子,须要用较强的碱才干把酯变成负离子.如异丁酸乙酯在三苯甲基钠感化下,可以进行缩合,而在乙醇钠感化下则不克不及产生反响:两种不合的酯也能产生酯缩合,理论上可得到四种不合的产品,称为混杂酯缩合,在制备上没有太大意义.假如个中一个酯分子中既无α-氢原子,并且烷氧羰基又比较生动时,则仅生成一种缩合产品.如苯甲酸酯.甲酸酯.草酸酯.碳酸酯等.与其它含α-氢原子的酯反响时,都只生成一种缩合产品.现实上这个反响不限于酯类自身的缩合,酯与含生动亚甲基的化合物都可以产生如许的缩合反响,这个反响可以用下列通式暗示:十三.Claisen—Schmidt 反响一个无氢原子的醛与一个带有氢原子的脂肪族醛或酮在稀氢氧化钠水溶液或醇溶液存鄙人产生缩合反响,并掉水得到不饱和醛或酮:反响机理反响实例十四.Claisen 重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚.当烯丙基芳基醚的两个邻位未被代替基占满时,重排重要得到邻位产品,两个邻位均被代替基占领时,重排得到对位产品.对位.邻位均被占满时不产生此类重排反响.交叉反响试验证实:Claisen重排是分子内的重排.采取 g-碳14C 标识表记标帜的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键产生位移.两个邻位都被代替的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连.反响机理Claisen 重排是个协同反响,中央经由一个环状过渡态,所以芳环上代替基的电子效应对重排无影响.从烯丙基芳基醚重排为邻烯丙基酚经由一次[3,3]s 迁徙和一次由酮式到烯醇式的互变异构;两个邻位都被代替基占领的烯丙基芳基酚重排时先经由一次[3,3]s 迁徙到邻位(Claisen 重排),因为邻位已被代替基占领,无法产生互变异构,接着又产生一次[3,3]s 迁徙()到对位,然后经互变异构得到对位烯丙基酚.代替的烯丙基芳基醚重排时,无论本来的烯丙基双键是Z-构型照样E-构型,重排后的新双键的构型都是E-型,这是因为重排反响所经由的六员环状过渡态具有稳固椅式构象的缘故.反响实例Claisen 重排具有广泛性,在醚类化合物中,假如消失烯丙氧基与碳碳相连的构造,就有可能产生Claisen 重排.十五.Clemmensen 还原醛类或酮类分子中的羰基被锌汞齐和浓盐酸还原为亚甲基:此法只实用于对酸稳固的化合物.对酸不稳固而对碱稳固的化合物可用还原.反响机理本反响的反响机理较庞杂,今朝尚不很清晰.反响实例十六.Combes 喹啉合成法Combes合成法是合成喹啉的另一种办法,是用芳胺与1,3-二羰基化合物反响,起首得到高产率的β-氨基烯酮,然后在浓硫酸感化下,羰基氧质子化后的羰基碳原子向氨基邻位的苯环碳原子进行亲电进攻,关环后,再脱水得到喹啉.反响机理在氨基的间位有强的邻.对位定位基团消失时,关环反响轻易产生;但当强邻.对位定位基团消失于氨基的对位时,则不轻易产生关环反响.反响实例十七.Cope 清除反响叔胺的N-氧化物(氧化叔胺)热解时生成烯烃和N,N-二代替羟胺,产率很高.现实上只需将叔胺与氧化剂放在一路,不需分别出氧化叔胺即可持续进行反响,例如在湿润的二甲亚砜或四氢呋喃中这个反响可在室温进行.此反响前提平和.副反响少,反响进程中不产生重排,可用来制备很多烯烃.当氧化叔胺的一个烃基上二个β位有氢原子消失时,清除得到的烯烃是混杂物,但是Hofmann产品为主;如得到的烯烃有顺反异构时,一般以 E-型为主.例如:反响机理这个反响是E2顺式清除反响,反响进程中形成一个平面的五员环过度态,氧化叔胺的氧作为进攻的碱:要产生如许的环状构造,氨基和β-氢原子必须处于统一侧,并且在形成五员环过度态时,α,β-碳原子上的原子基团呈重叠型,如许的过度态须要较高的活化能,形成后也很不稳固,易于进行清除反响.反响实例十八.Cope 重排1,5-二烯类化合物受热时产生类似于 O-烯丙基重排为 C-烯丙基的重排反响()反响称为Cope重排.这个反响30多年来引起人们的广泛留意.1,5-二烯在150—200℃单独加热短时光就轻易产生重排,并且产率异常好.Cope重排属于周环反响,它和其它周环反响的特色一样,具有高度的立体选择性.例如:内消旋-3,4-二甲基-1,5-己二烯重排后,得到的产品几乎全体是(Z, E)-2,6辛二烯:反响机理Cope重排是[3,3]s-迁徙反响,反响进程是经由一个环状过渡态进行的协同反响:在立体化学上,表示为经由椅式环状过渡态:反响实例十九.Curtius 反响酰基叠氮化物在惰性溶剂中加热分化生成异氰酸酯:异氰酸酯水解则得到胺:反响机理反响实例二十.Crigee,R 反响1,2-二元醇类的氧化产品因所用的氧化剂的种类而不合.用K2Cr2O7或KMnO4氧化时生成酸类.用特别氧化剂四乙醋酸铅在CH3COOH或苯等不生动有机溶剂中缓和氧化,生成二分子羰基化合物(醛或酮).氧化反响也可以在酸催化剂(三氯醋酸)存鄙人进行.本反响被广泛地运用于研讨醇类构造及制备醛.酮类,产率很高.反响机理反响进程中师长教师成环酯中央产品,进一步C--C键裂开成醛或酮.酸催化的场合,反响过程可以用下式暗示:反响实例二十一.Dakin 反响酚醛或酚酮类用H2O2在NaOH存鄙人氧化时,可将分子中的-CHO基或CH3CO-基被-OH基所置换,生成相对应的酚类.本反响可运用以制备多远酚类.反响机理反响实例二十二.Elbs 反响羰基的邻位有甲基或亚甲基的二芳基酮,加热时产生环化脱氢感化,生成蒽的衍生物:因为这个反响平日是在回流温度或高达400-450 °C的温度规模内进行,不必催化剂和溶剂,直到反响物没有水放出为止,在如许的高温前提下,一部分原料和产品产生碳化,部分原料酮被释放出的水所裂解,烃基产生清除或降解以及分子重排等副反响,致使产率不高.反响机理本反响的机理尚不清晰.反响实例二十三.Edvhweiler-Clarke 反响在过量甲酸存鄙人,一级胺或二级胺与甲醛反响,得到甲基化后的三级胺:甲醛在这里作为一个甲基化试剂.反响机理反响实例二十四.将一元酚类或类似化合物用过硫酸钾在碱性溶液中氧化羟基引入在原有羟基的对位或邻位,生成二元酚类.分子中的醛基或双键等都不影响.产率约20~48%.过硫酸钾的水溶液在加热时放出氧:芳伯胺类如用本试剂氧化时,变成硝基化合物.反响机理反响实例二十五.Favorskii 重排a-卤代酮在氢氧化钠水溶液中加热重排生成含雷同碳原子数的羧酸;如为环状a-卤代酮,则导致环缩小.如用醇钠的醇溶液,则得羧酸酯:此法可用于合成张力较大的四员环.反响机理反响实例二十六.Friedel-Crafts 烷基化反响芳烃与卤代烃.醇类或烯类化合物在Lewis催化剂(如AlCl3,FeCl3,H2SO4, H3PO4, BF3, HF等)存鄙人,产生芳环的烷基化反响.卤代烃反响的生动性次序为:RF > RCl > RBr > RI ; 当烃基超出3个碳原子时,反响进程中易产生重排.反响机理起首是卤代烃.醇或烯烃与催化剂如三氯化铝感化形成碳正离子:所形成的碳正离子可能产生重排,得到较稳固的碳正离子:碳正离子作为亲电试剂进攻芳环形成中央体s-络合物,然后掉去一个质子得到产生亲电代替产品:反响实例二十七.Friedel-Crafts酰基化反响芳烃与酰基化试剂如酰卤.酸酐.羧酸.烯酮等在Lewis酸(通经常运用无水三氯化铝)催化下产生酰基化反响,得到芬芳酮:这是制备芬芳酮类最重要的办法之一,在酰基化中不产生烃基的重排.反响机理反响实例二十八.Fries 重排酚酯在Lewis酸存鄙人加热,可产生酰基重排反响,生成邻羟基和对羟基芳酮的混杂物.重排可以在硝基苯.硝基甲烷等溶剂中进行,也可以不必溶剂直接加热进行.邻.对位产品的比例取决于酚酯的构造.反响前提和催化剂等.例如,用多聚磷酸催化时重要生成对位重排产品,而用四氯化钛催化时则重要生成邻位重排产品.反响温度对邻.对位产品比例的影响比较大,一般来讲,较低温度(如室温)下重排有利于形成对位异构产品(动力学掌握),较高温度下重排有利于形成邻位异构产品(热力学掌握).反响机理反响实例二十九.Fischer,O-Hepp,E 重排N-亚硝基芳胺用盐酸或氢溴酸或其乙醇溶液处理时氨基氮上的亚硝基转移到芳核上去形成p-亚硝基芳胺(对位重排):平日产生对位重排,但在奈系化合物中如N-亚硝基-N-加基-2-奈胺则产生邻位重排成1-亚硝基化合物:反响机理在HCl存鄙人,N-亚硝基化合物起首解离成仲胺及NOCl然落后行亚硝基化:三十.Gabriel 合成法邻苯二甲酰亚胺与氢氧化钾的乙醇溶液感化改变成邻苯二甲酰亚胺盐,此盐和卤代烷反响生成N-烷基邻苯二甲酰亚胺,然后在酸性或碱性前提下水解得到一级胺和邻苯二甲酸,这是制备纯净的一级胺的一种办法.有些情形下水解很艰苦,可以用肼解来代替:反响机理邻苯二甲酰亚胺盐和卤代烷的反响是亲核代替反响,代替反响产品的水解进程与酰胺的水解类似.反响实例三十一.Gattermann 反响重氮盐用新制的铜粉代替亚铜盐(见)作催化剂,与浓盐酸或氢溴酸产生置换反响得到氯代或溴代芳烃:本法长处是操纵比较简略,反响可在较低温度下进行,缺陷是其产率一般较低.反响机理见反响实例三十二.Gattermann-Koch 反响芬芳烃与等分子的一氧化碳及氯化氢气体在加压和催化剂(三氯化铝及氯化亚铜)存鄙人反响,生成芬芳醛:反响机理反响实例三十三.Gomberg-Bachmann 反响芬芳重氮盐在碱性前提下与其它芬芳族化合物偶联生成联苯或联苯衍生物:反响机理反响实例三十四.Hantzsch 合成法两分子b-羰基酸酯和一分子醛及一分子氨产生缩合反响,得到二氢吡啶衍生物,再用氧化剂氧化得到吡啶衍生物.这是一个很广泛的反响,用于合成吡啶同系物.反响机理反响进程可能是一分子b-羰基酸酯和醛反响,另一分子b-羰基酸酯和氨反响生成b-氨基烯酸酯,所生成的这两个化合物再产生Micheal加成反响,然后掉水关环生成二氢吡啶衍生物,它很溶液脱氢而芳构化,例如用亚硝酸或铁氰化钾氧化得到吡啶衍生物:三十五.Haworth 反响萘和丁二酸酐产生然后按尺度的办法还原.关环.还原.脱氢得到多环芬芳族化合物.反响机理见反响实例三十六.Hell-Volhard-Zelinski 反响羧酸在催化量的三卤化磷或红磷感化下,能与卤素产生a-卤代反响生成a-卤代酸:本反响也可以用酰卤作催化剂.反响机理反响实例三十七.Hinsberg 反响伯胺.仲胺分别与对甲苯磺酰氯感化生成响应的对甲苯磺酰胺沉淀,个中伯胺生成的沉淀能溶于碱(如氢氧化钠)溶液,仲胺生成的沉淀则不溶,叔胺与对甲苯磺酰氯不反响.此反响可用于伯仲叔胺的分别与判定.三十八.Hofmann 烷基化卤代烷与氨或胺产生烷基化反响,生成脂肪族胺类:因为生成的伯胺亲核性平日比氨强,能持续与卤代烃反响,是以本反响不成防止地产生仲胺.叔胺和季铵盐,最后得到的往往是多种产品的混杂物.用大过量的氨可防止多代替反响的产生,从而可得到优越产率的伯胺.反响机理反响为典范的亲核代替反响(S N1或S N2)三十九.Hofmann 清除反响季铵碱在加热前提下(100--200°C)产生热分化,当季铵碱的四个烃基都是甲基时,热分化得到甲醇和三甲胺:假如季铵碱的四个烃基不合,则热分化时老是得到含代替基起码的烯烃和叔胺:反响实例四十.Hofmann 重排(降解)酰胺用溴(或氯)在碱性前提下处理改变成少一个碳原子的伯胺:反响机理反响实例四十一.Houben-Hoesch 反响酚或酚醚在氯化氢和氯化锌等Lewis酸的存鄙人,与腈感化,随落后行水解,得到酰基酚或酰基酚醚:反响机理反响机理较庞杂,今朝尚未完整解释反响实例。


大学化学有机化学的基本反应与机理一、醇的脱水反应及机理醇是一类含有羟基(-OH)官能团的有机化合物。
在一定条件下,醇可以发生脱水反应,即羟基脱除一个氢原子和一个氧原子形成水,同时生成一个烯烃。
这种反应常见于工业生产中或实验室合成有机化合物的过程。
脱水反应的机理可以分为酸催化和碱催化两种情况。
1. 酸催化的脱水反应机理通常情况下,酸催化的脱水反应发生在较高温度下。
以乙醇为例,其脱水反应可以被硫酸作为催化剂催化。
催化剂中的硫酸可以负责将醇中的羟基质子化形成羟基阳离子,从而增强羟基上的电荷密度。
这使得羟基上的氧原子更容易离去,形成水分子。
同时,形成的乙烯基阳离子会接受甲基的亲电攻击,并且从硫酸中再生出硫酸。
2. 碱催化的脱水反应机理碱催化的脱水反应在较低温度下进行。
以乙醇为例,可以用氢氧化钠作为碱的催化剂。
催化剂中的氢氧化钠会负责从醇中去质子化羟基,形成氧化钠离子和醇的负氧离子。
羟基上的负氧离子很容易离去,形成水分子。
形成的烯烃则会与氢氧化钠反应,再生出氢氧化钠。
二、醛和酮的加成反应及机理醛和酮是羰基化合物,其分子中含有羰基(C=O)官能团。
在一定条件下,醛和酮可以发生加成反应,即通过在羰基碳上加入其他官能团的方式合成新的化合物。
醛和酮的加成反应可以分为亲核加成和胺催化的亲核加成两种情况。
1. 亲核加成的机理亲核加成是指在醛或酮的羰基碳上发生亲核攻击,将一个亲核试剂加到羰基碳上,并形成一条新的键。
以乙醛和氨水的加成反应为例,氨水中的氨为亲核试剂,而水分子则是副产物。
该反应通过羰基碳上的部分正电荷互相吸引,在亲核试剂的攻击下,羰基碳上的δ+位点与亲核试剂上的亲核试对形成新的共价键。
2. 胺催化的亲核加成的机理胺催化的亲核加成是指胺作为催化剂,在亲核试剂作用下催化醛或酮的加成。
以戊二酮和苯胺的加成反应为例,苯胺作为催化剂参与了反应过程。
首先,苯胺与戊二酮发生氢键形成氮质子化胺。
然后,产生的胺质子对氨基负离子进一步质子化,形成羟胺离子。

常见的有机反应机理Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
ﻫ本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷R'X 的烷基和亚磷酸三烷基酯 (RO)3P的烷基相同(即 R' = R),则Arbuzov反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理一般认为是按SN2 进行的分子内重排反应:反应实例Arndt-Eister反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例Baeyer----Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。

Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则 Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理一般认为是按 SN2 进行的分子内重排反应:反应实例Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例Baeyer----Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。

有机化学反应机理+范例+原理1. Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例2.Baeyer----Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。
3.Beckmann 重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例4.Birch 还原芳香化合物用碱金属(钠、钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇)的混合液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物。
反应机理首先是钠和液氨作用生成溶剂化点子,然后苯得到一个电子生成自由基负离子(Ⅰ),这是苯环的л电子体系中有7个电子,加到苯环上那个电子处在苯环分子轨道的反键轨道上,自由基负离子仍是个环状共轭体系,(Ⅰ)表示的是部分共振式。
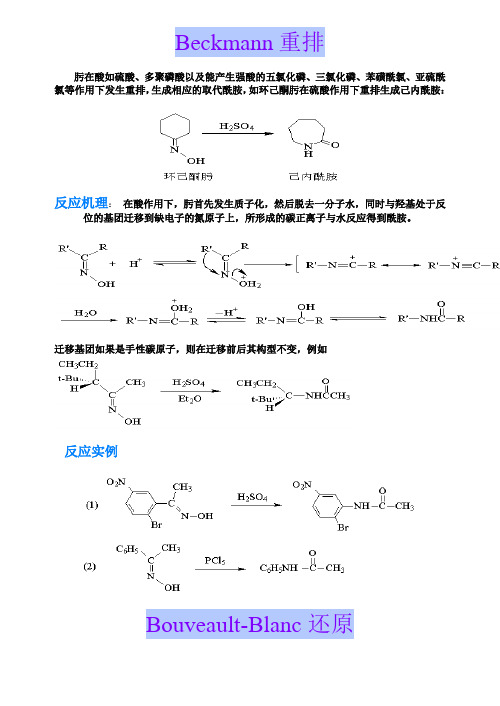
Beckmann重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理:在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如反应实例Bouveault-Blanc还原反应机理反应实例Claisen-Schmidt反应一个无α-氢原子的醛与一个带有α-氢原子的脂肪族醛或酮在稀氢氧化钠水溶液或醇溶液存在下发生缩合反应,并失水得到α,β-不饱和醛或酮:反应机理反应实例Claisen酯缩合反应二元羧酸酯的分子内酯缩合见Dieckmann 缩合反应。
反应机理:反应实例:Cope 消除反应反应机理反应实例Cope重排1,5-二烯类化合物受热时发生类似于O-烯丙基重排为C-烯丙基的重排反应(Claisen重排)反应称为Cope重排。
这个反应30多年来引起人们的广泛注意。
1,5-二烯在150—200℃单独加热短时间就容易发生重排,并且产率非常好。
Cope重排属于周环反应,它和其它周环反应的特点一样,具有高度的立体选择性。
例如:内消旋-3,4-二甲基-1,5-己二烯重排后,得到的产物几乎全部是(Z, E)-2,6辛二烯:反应机理Cope重排是[3,3] -迁移反应,反应过程是经过一个环状过渡态进行的协同反应:在立体化学上,表现为经过椅式环状过渡态:反应实例Clemmensen还原醛类或酮类分子中的羰基被锌汞齐和浓盐酸还原为亚甲基:此法只适用于对酸稳定的化合物。
对酸不稳定而对碱稳定的化合物可用Wolff-Kishner-黄鸣龙反应还原。
反应实例Diels-Alder反应含有一个活泼的双键或叁键的化合物(亲双烯体)与共轭二烯类化合物(双烯体)发生1,4-加成,生成六员环状化合物:这个反应极易进行并且反应速度快,应用范围极广泛,是合成环状化合物的一个非常重要的方法。

常见的有机反应机理Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则 Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理一般认为是按 S N2 进行的分子内重排反应:反应实例Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例Baeyer----Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。


有机化学人名反应机理(比较完整)1.arbuzov反应卤代烷反应时,其活性次序为:r'i>r'br>r'cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a-或b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应就是由醇制取卤代烷的较好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制取:一般认为是按sn2进行的分子内重排反应:2.arndt-eister反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
重氮甲烷与酰氯反应首先构成重氮酮(1),(1)在氧化银催化剂下与水共热,获得酰基卡宾(2),(2)出现重排得烯酮(3),(3)与水反应分解成酸,若与醇或氨(胺)反应,则得酯或酰胺。
3.baeyer----villiger反应过酸先与羰基展开亲核差率,然后酮羰基上的一个烃基带着一对电子搬迁至-o-o-基团中与羰基碳原子轻易相连的氧原子上,同时出现o-o键异裂。
因此,这就是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不等距的酮水解时,在重排步骤中,两个基团均可搬迁,但是还是存有一定的选择性,按搬迁能力其顺序为:4.beckmann重排肟在酸例如硫酸、磷酸酯磷酸以及能够产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等促进作用下出现重排,分解成适当的替代酰胺,例如环己酮肟在硫酸促进作用下重排分解成己内酰胺:在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
搬迁基团如果就是手性碳原子,则在搬迁前后其构型维持不变。
5.bouveault---blanc还原脂肪族羧酸酯需用金属钠和醇还原成得一级醇。
α,β-不饱和羧酸酯还原成得适当的饱和状态醇。
芳香酸酯也可以展开本反应,但收率较低。

有机化学反应机理总结(较全)有机化学反应机理总结 (完整版)本文总结了几种常见的有机化学反应的机理,并提供了相关的示意图。
以期帮助读者更好地理解有机化学反应的机理和反应过程。
1. 反应类型1: 取代反应取代反应是有机化学中最基本的反应类型之一。
它涉及到一个分子或它的一部分被另一个原子或基团取代的过程。
以下是一个典型的取代反应的机理示意图:机理步骤:1. 亲核试剂与底物发生反应,亲核试剂攻击底物的部分阳离子或电子不足的原子。
2. 形成一个中间体,中间体中的某个基团离开。
3. 离开基团被亲核试剂取代,形成最终产物。
2. 反应类型2: 加成反应加成反应发生在两个分子之间,它们在反应中结合形成一个新的分子。
加成反应的机理示意图如下所示:机理步骤:1. 两个反应物中的亲核试剂和电荷不足的物种发生相互作用。
2. 形成一个键合物中间体。
3. 中间体通过质子转移或亲核试剂攻击等步骤,产生最终产物。
3. 反应类型3: 消除反应消除反应是一种从底物中除去一些原子或基团的反应,生成了双键或环。
以下是消除反应的机理示意图:机理步骤:1. 底物中的一个基团被移除,形成一个中间体。
2. 中间体中的某个原子或基团与另一个原子或基团形成新的共价键。
3. 生成最终产物。
以上是几种常见有机化学反应的机理总结。
希望本文能对读者理解有机化学反应的机理和反应过程有所帮助。
参考文献:请注意,以上内容仅供参考,具体反应机理可能会因具体情况而有所不同。

化学中的有机合成反应原理及机理有机化学是化学的分支,主要研究有机物,即碳氢化合物及其衍生物。
在有机合成中,合成反应是最基本的实验操作之一,也是实现有机分子结构设计和构建的关键。
有机合成反应原理有机合成反应原理可以大致分为三类:加成反应、消除反应和取代反应。
1、加成反应(Addition Reaction)加成反应是指在化合物中两个原子团之间发生相互作用,形成一个新的化学键,通常产生了对于原有分子来说更大的分子量。
加成反应是有机化学最基本、最常见的反应类型之一,主要包括π键的加成反应和偶极加成反应。
(1)π键的加成反应π键的加成反应是指当烯烃与其他原子团相遇时,它们之间的π键可以发生开裂,两个不饱和的单元分别与加成的原子团结合,形成一个新的化合物。
例如,乙烯与氢气反应生成乙烷,如下所示:C2H4 + H2 → C2H6(2)偶极加成反应偶极加成反应是指存在偶极矩的化合物与另一个带有相反偶极矩的化学物质结合,形成键合物,且偶极矩消失。
例如,醛或酮与硫酸铵反应,生成席夫酸盐。
RCOR' + NH4HSO4 → RCOOCH3 + H2SO4 + NH32、消除反应(Elimination Reaction)消除反应是指某个分子中的一个基团离开后,该分子的反应物结构发生变化。
例如,醇在酸性溶液中加热,可以进行脱水反应。
R-OH → R-OH2+ → R+ + H2O3、取代反应(Substitution Reaction)取代反应是有机化学中最基本的反应类型之一,指一种化合物中的原子团或基团被另一种原子或基团所取代的反应。
取代反应可以分为有机物中的芳香取代反应和脂肪族烷基取代反应。
(1)芳香取代反应芳香取代反应是指原有芳环中的氢原子被取代或加成另一个基,通常反应发生在带有空位的或能通过羟基、氨基、羟基苯甲酸等配体引发的机制中,如下所示:C6H6 + Cl2 → C6H5Cl + HCl(2)脂肪族烷基取代反应脂肪族烷基取代反应是指有机物中的烷基或类似物中的某个氢原子被取代或加成另一个基团的反应,通常发生在角化反应中。

基础有机化学反应总结一、烯烃1、卤化氢加成 (1)CHCH 2RHXCH 3RX【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。
【机理】CH 2CH 3+CH 3CH 3X +CH 3CH 3+H +CH 2+C3X +CH 3X主次【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。
【注】碳正离子的重排 (2)CHCH 2RCH 2CH 2R BrHBrROOR【特点】反马氏规则 【机理】 自由基机理(略)【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。
【本质】不对称烯烃加成时生成稳定的自由基中间体。
【例】CH 2CH3BrCH CH 2BrC H 3CH +CH 3C H 3HBrBrCH 3CH 2CH 2BrCH CH 3C H 32、硼氢化—氧化CHCH 2R CH 2CH 2R OH1)B 2H 62)H 2O 2/OH-【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。
【机理】2CH 33H 323H 32CH CH 2CH 32CH CH=CH (CH 3CH 2CH 2)3-H 3CH 2CH 2C22CH 3CH 2OCH 2CH 2CH 3H 3CH 2CH 2C2CH 2CH 3+OH -OHB-OC H 2CH 2CH 3CH 2CH 2CH 3H 3CH 2CH 2BOC H 2CH 2CH 3CH 2CH 2CH 3H 2CH 2CH 3HOO -B(OCH 2CH 2CH 3)3B(OCH 2CH 2CH 3)3+3NaOH 3NaOH3HOC H 2CH 2CH 33+Na 3BO 32【例】CH 31)BH 32)H 2O 2/OH -CH 3HH OH3、X 2加成C CBr /CCl CC Br【机理】CC CC Br BrC CBr +CC Br OH 2+-H +CC Br OH【注】通过机理可以看出,反应先形成三元环的溴鎓正离子,然后亲和试剂进攻从背面进攻,不难看出是反式加成。

有机化学反应机理一、引言有机化学反应机理是研究有机化合物在反应过程中发生的分子转化和反应速率的原理和规律的科学。
它对于揭示有机反应的本质、预测反应产物和优化反应条件具有重要意义。
本文将以几种常见有机化学反应为例,介绍其反应机理及相关特点。
二、酯化反应酯化反应是有机化学中一种重要的酸催化反应。
它通过酸催化剂使酯酸酐与醇发生取代反应,生成酯和水。
酸催化剂通常是质子酸,如硫酸、磷酸等。
反应机理包括亲核进攻、质子化、质子转移和亲核消除等步骤。
该反应机理的研究可以为酯化反应条件的优化和产物选择提供理论依据。
三、氧化反应氧化反应是有机化学中常见的重要反应类型之一。
它通过氧化剂使有机物中的氢原子被氧原子取代,生成相应的氧化产物。
氧化反应的机理复杂,常涉及自由基、电子转移和氧化还原等过程。
例如,醇的氧化常用氧气或过氧化氢作为氧化剂,生成相应的醛或酮。
氧化反应机理的研究可以为氧化反应条件的控制和产物选择提供理论指导。
四、加成反应加成反应是有机化学中一类重要的反应类型,指两个或多个反应物中的原子团通过共价键形成新的化学键。
加成反应的机理多样,常见的有电子亲和性反应、亲核性反应、自由基反应等。
例如,醛和酮与亲核试剂发生加成反应,生成相应的醇或酮。
加成反应机理的研究可以为反应条件的优化和产物选择提供理论支持。
五、消除反应消除反应是有机化学中一种重要的反应类型,指通过断裂一个碳-碳键和一个碳-氢键,生成一个新的双键或三键。
消除反应的机理多样,常见的有β-消除、酸催化消除、碱催化消除等。
例如,卤代烷和碱发生消除反应,生成烯烃。
消除反应机理的研究可以为反应条件的控制和产物选择提供理论指导。
六、总结有机化学反应机理的研究对于理解有机反应的本质、预测反应产物和优化反应条件具有重要意义。
本文以酯化反应、氧化反应、加成反应和消除反应为例,介绍了它们的反应机理及相关特点。
希望通过对这些反应机理的了解,能够提高我们对有机化学反应的理解和应用能力。

有机化学反应及其反应原理有机化学反应是指有机化合物之间发生的化学变化过程。
有机化学反应广泛应用于有机合成、药物制备、材料合成等领域。
本文将介绍几种常见的有机化学反应及其反应原理。
一、酯化反应酯化反应是指酸与醇在酸催化下发生的反应,生成酯和水。
酯化反应常用于酯的合成。
反应原理是酸催化下,酸与醇发生质子转移,生成酯。
酯化反应通常需要使用酸催化剂,如硫酸、磷酸等。
二、加成反应加成反应是指两个或多个有机化合物分子在一定条件下发生化学键的形成,生成新的化合物。
加成反应常用于合成有机化合物。
反应原理是通过共价键的形成,使反应物中的原子重新组合,生成新的化合物。
加成反应种类繁多,常见的有烯烃的加成反应、醛酮的加成反应等。
三、取代反应取代反应是指有机化合物中的一个官能团被另一个官能团取代的反应。
取代反应常用于合成新的有机化合物。
反应原理是通过亲核试剂或电子试剂与反应物发生反应,将一个官能团取代为另一个官能团。
取代反应种类繁多,常见的有烷基取代反应、卤代取代反应等。
四、氧化反应氧化反应是指有机化合物中的氢原子被氧原子取代的反应。
氧化反应常用于氧化剂的应用以及有机物的功能化改造。
反应原理是氧化剂与反应物中的氢原子发生反应,生成相应的氧化产物。
氧化反应种类繁多,常见的有醇的氧化反应、醛的氧化反应等。
五、还原反应还原反应是指有机化合物中的氧原子被氢原子取代的反应。
还原反应常用于还原剂的应用以及有机物的还原改造。
反应原理是还原剂与反应物中的氧原子发生反应,生成相应的还原产物。
还原反应种类繁多,常见的有酮的还原反应、羰基的还原反应等。
六、缩合反应缩合反应是指两个分子中的官能团发生反应,生成一个分子的反应。
缩合反应常用于合成大分子化合物或环状化合物。
反应原理是官能团之间发生反应,形成新的化学键,使反应物分子数减少。
缩合反应种类繁多,常见的有醛酮的缩合反应、胺的缩合反应等。
总结起来,有机化学反应涵盖了酯化反应、加成反应、取代反应、氧化反应、还原反应和缩合反应等多种类型。
1.A rndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例2.Baeyer----Villiger 反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。
3.Beckmann 重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例4.Birch还原芳香化合物用碱金属(钠、钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇)的混合液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物。
反应机理首先是钠和液氨作用生成溶剂化点子,然后苯得到一个电子生成自由基负离子(Ⅰ),这是苯环的л电子体系中有7个电子,加到苯环上那个电子处在苯环分子轨道的反键轨道上,自由基负离子仍是个环状共轭体系,(Ⅰ)表示的是部分共振式。
(Ⅰ)不稳定而被质子化,随即从乙醇中夺取一个质子生成环己二烯自由基(Ⅱ)。
(Ⅱ)在取得一个溶剂化电子转变成环己二烯负离子(Ⅲ),(Ⅲ)是一个强碱,迅速再从乙醇中夺取一个电子生成1,4-环己二烯。
环己二烯负离子(Ⅲ)在共轭链的中间碳原子上质子化比末端碳原子上质子快,原因尚不清楚。
反应实例取代的苯也能发生还原,并且通过得到单一的还原产物。
例如5.Bouveault---Blanc 还原脂肪族羧酸酯可用金属钠和醇还原得一级醇。
α,β-不饱和羧酸酯还原得相应的饱和醇。
芳香酸酯也可进行本反应,但收率较低。
本法在氢化锂铝还原酯的方法发现以前,广泛地被使用,非共轭的双键可不受影响。
反应机理首先酯从金属钠获得一个电子还原为自由基负离子,然后从醇中夺取一个质子转变为自由基,再从钠得一个电子生成负离子,消除烷氧基成为醛,醛再经过相同的步骤还原成钠,再酸化得到相应的醇。
反应实例醛酮也可以用本法还原,得到相应的醇:6.Cannizzaro 反应凡α位碳原子上无活泼氢的醛类和浓NaOH或KOH水或醇溶液作用时,不发生醇醛缩合或树脂化作用而起歧化反应生成与醛相当的酸(成盐)及醇的混合物。
此反应的特征是醛自身同时发生氧化及还原作用,一分子被氧化成酸的盐,另一分子被还原成醇:脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会发生此反应,其他醛类与强碱液,作用发生醇醛缩合或进一步变成树脂状物质。
具有α-活泼氢原子的醛和甲醛首先发生羟醛缩合反应,得到无α-活泼氢原子的β-羟基醛,然后再与甲醛进行交叉Cannizzaro反应,如乙醛和甲醛反应得到季戊四醇:反应机理醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢负离子的形式转移到另一分子的羰基不能碳原子上。
反应实例7.Claisen 酯缩合反应含有α-氢的酯在醇钠等碱性缩合剂作用下发生缩合作用,失去一分子醇得到β-酮酸酯。
如2分子乙酸乙酯在金属钠和少量乙醇作用下发生缩合得到乙酰乙酸乙酯。
二元羧酸酯的分子内酯缩合见Dieckmann缩合反应。
反应机理乙酸乙酯的α-氢酸性很弱(pK a-24.5),而乙醇钠又是一个相对较弱的碱(乙醇的pK a~15.9),因此,乙酸乙酯与乙醇钠作用所形成的负离子在平衡体系是很少的。
但由于最后产物乙酰乙酸乙酯是一个比较强的酸,能与乙醇钠作用形成稳定的负离子,从而使平衡朝产物方向移动。
所以,尽管反应体系中的乙酸乙酯负离子浓度很低,但一形成后,就不断地反应,结果反应还是可以顺利完成。
常用的碱性缩合剂除乙醇钠外,还有叔丁醇钾、叔丁醇钠、氢化钾、氢化钠、三苯甲基钠、二异丙氨基锂(LDA)和Grignard试剂等。
反应实例如果酯的α-碳上只有一个氢原子,由于酸性太弱,用乙醇钠难于形成负离子,需要用较强的碱才能把酯变为负离子。
如异丁酸乙酯在三苯甲基钠作用下,可以进行缩合,而在乙醇钠作用下则不能发生反应:两种不同的酯也能发生酯缩合,理论上可得到四种不同的产物,称为混合酯缩合,在制备上没有太大意义。
如果其中一个酯分子中既无α-氢原子,而且烷氧羰基又比较活泼时,则仅生成一种缩合产物。
如苯甲酸酯、甲酸酯、草酸酯、碳酸酯等。
与其它含α-氢原子的酯反应时,都只生成一种缩合产物。
实际上这个反应不限于酯类自身的缩合,酯与含活泼亚甲基的化合物都可以发生这样的缩合反应,这个反应可以用下列通式表示:8.Claisen—Schmidt 反应一个无α-氢原子的醛与一个带有α-氢原子的脂肪族醛或酮在稀氢氧化钠水溶液或醇溶液存在下发生缩合反应,并失水得到α,β-不饱和醛或酮:反应机理反应实例9.Claisen 重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。
当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。
对位、邻位均被占满时不发生此类重排反应。
交叉反应实验证明:Claisen重排是分子内的重排。
采用 g-碳 14C 标记的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键发生位移。
两个邻位都被取代的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连。
反应机理Claisen 重排是个协同反应,中间经过一个环状过渡态,所以芳环上取代基的电子效应对重排无影响。
从烯丙基芳基醚重排为邻烯丙基酚经过一次[3,3]s 迁移和一次由酮式到烯醇式的互变异构;两个邻位都被取代基占据的烯丙基芳基酚重排时先经过一次[3,3]s 迁移到邻位(Claisen 重排),由于邻位已被取代基占据,无法发生互变异构,接着又发生一次[3,3]s 迁移(Cope 重排)到对位,然后经互变异构得到对位烯丙基酚。
取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是Z-构型还是E-构型,重排后的新双键的构型都是E-型,这是因为重排反应所经过的六员环状过渡态具有稳定椅式构象的缘故。
反应实例Claisen 重排具有普遍性,在醚类化合物中,如果存在烯丙氧基与碳碳相连的结构,就有可能发生Claisen 重排。
10.Clemmensen 还原醛类或酮类分子中的羰基被锌汞齐和浓盐酸还原为亚甲基:此法只适用于对酸稳定的化合物。
对酸不稳定而对碱稳定的化合物可用Wolff-Kishner-黄鸣龙反应还原。
反应机理本反应的反应机理较复杂,目前尚不很清楚。
反应实例11.Cope 消除反应叔胺的N-氧化物(氧化叔胺)热解时生成烯烃和N,N-二取代羟胺,产率很高。
实际上只需将叔胺与氧化剂放在一起,不需分离出氧化叔胺即可继续进行反应,例如在干燥的二甲亚砜或四氢呋喃中这个反应可在室温进行。
此反应条件温和、副反应少,反应过程中不发生重排,可用来制备许多烯烃。
当氧化叔胺的一个烃基上二个β位有氢原子存在时,消除得到的烯烃是混合物,但是Hofmann产物为主;如得到的烯烃有顺反异构时,一般以E-型为主。
例如:反应机理这个反应是E2顺式消除反应,反应过程中形成一个平面的五员环过度态,氧化叔胺的氧作为进攻的碱:要产生这样的环状结构,氨基和β-氢原子必须处于同一侧,并且在形成五员环过度态时,α,β-碳原子上的原子基团呈重叠型,这样的过度态需要较高的活化能,形成后也很不稳定,易于进行消除反应。
反应实例12.Cope 重排1,5-二烯类化合物受热时发生类似于 O-烯丙基重排为 C-烯丙基的重排反应(Claisen 重排)反应称为Cope重排。
这个反应30多年来引起人们的广泛注意。
1,5-二烯在150—200℃单独加热短时间就容易发生重排,并且产率非常好。
Cope重排属于周环反应,它和其它周环反应的特点一样,具有高度的立体选择性。
例如:内消旋-3,4-二甲基-1,5-己二烯重排后,得到的产物几乎全部是(Z, E)-2,6辛二烯:反应机理Cope重排是[3,3]s-迁移反应,反应过程是经过一个环状过渡态进行的协同反应:在立体化学上,表现为经过椅式环状过渡态:反应实例13.Friedel-Crafts 烷基化反应芳烃与卤代烃、醇类或烯类化合物在Lewis催化剂(如AlCl3,FeCl3, H2SO4, H3PO4, BF3, HF 等)存在下,发生芳环的烷基化反应。
卤代烃反应的活泼性顺序为:RF > RCl > RBr > RI ; 当烃基超过3个碳原子时,反应过程中易发生重排。
反应机理首先是卤代烃、醇或烯烃与催化剂如三氯化铝作用形成碳正离子:所形成的碳正离子可能发生重排,得到较稳定的碳正离子:碳正离子作为亲电试剂进攻芳环形成中间体s-络合物,然后失去一个质子得到发生亲电取代产物:反应实例14.Friedel-Crafts 酰基化反应芳烃与酰基化试剂如酰卤、酸酐、羧酸、烯酮等在Lewis酸(通常用无水三氯化铝)催化下发生酰基化反应,得到芳香酮:这是制备芳香酮类最重要的方法之一,在酰基化中不发生烃基的重排。
反应机理反应实例15.Gattermann-Koch 反应芳香烃与等分子的一氧化碳及氯化氢气体在加压和催化剂(三氯化铝及氯化亚铜)存在下反应,生成芳香醛:反应机理反应实例16.Hell-Volhard-Zelinski 反应羧酸在催化量的三卤化磷或红磷作用下,能与卤素发生a-卤代反应生成a-卤代酸:本反应也可以用酰卤作催化剂。
反应机理反应实例17.Hinsberg 反应伯胺、仲胺分别与对甲苯磺酰氯作用生成相应的对甲苯磺酰胺沉淀,其中伯胺生成的沉淀能溶于碱(如氢氧化钠)溶液,仲胺生成的沉淀则不溶,叔胺与对甲苯磺酰氯不反应。
此反应可用于伯仲叔胺的分离与鉴定。
18.Hofmann 烷基化卤代烷与氨或胺发生烷基化反应,生成脂肪族胺类:由于生成的伯胺亲核性通常比氨强,能继续与卤代烃反应,因此本反应不可避免地产生仲胺、叔胺和季铵盐,最后得到的往往是多种产物的混合物。