DNA测序常见问题分析及解决办法总结
- 格式:doc
- 大小:71.00 KB
- 文档页数:2

DNA测序过程可能遇到的问题及分析对于一些生物测序公司(如Invitrogen等),我们的菌液或质粒经过PCR和酶切鉴定都没问题,但几天后的测序结果却无法另人满意。
为什么呢?PCR产物直接进行测序,在PCR产物长度以后将无反应信号,机器将产生许多N值。
这是由于Taq酶能够在PCR反应的末端非特异性地加上一个A碱基,我们所用的T载体克隆PCR产物就是应用该原理,通常PCR产物结束的位点,PCR产物测序一般末端的一个碱基为A(绿峰),也就是双脱氧核甘酸ddNTP终止反应的位置之前的A,A后的信号会迅速减弱。
N值情况一般是由于有未去除的染料单体造成的干扰峰。
该干扰峰和正常序列峰重叠在一起,有时机器377以下的测序仪无法正确判断出为何碱基。
有时,在序列的起始端的小片段容易丢失,导致起始区信号过低,机器有时也无法正确判读。
在序列的3’端易产生N值。
一个测序反应一般可以读出900bp以上的碱基(ABI3730可以达到1200bp),但是,只有一般600bp以前的碱基是可靠的,理想条件下,多至700bp的碱基都是可以用的。
一般在650bp以后的序列,由于测序毛细管胶的分辩率问题,会有许多碱基分不开,就会产生N值。
测序模板本身含杂合序列,该情况主要发生在PCR产物直接测序,由于PCR产物本身有突变或含等位基因,会造成在某些位置上有重叠峰,产生N值。
这种情况很容易判断,那就是整个序列信号都非常好,只有在个别位置有明显的重叠峰,视杂合度不同N值也不同。
测序列是从引物3’末端后第一个碱基开始的,所以就看不到引物序列。
有两种方法可以得到引物序列。
1.对于较短的PCR产物(<600bp),可以用另一端的引物进行测序,从另一端测序可以一直测通,可以在序列的末端得到该引物的反向互补序列。
对于较长的序列,一个测序反应测不通,就只能将PCR产物片段克隆到载体中,用载体上的通用引物(T7/SP6)进行测序。
载体上的通用引物与所插入序列间有一段距离,因此就可以得到完整的引物序列。
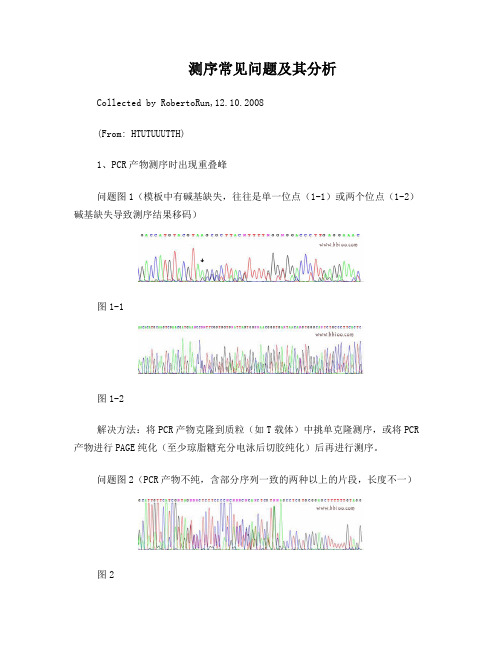
测序常见问题及其分析Collected by RobertoRun,12.10.2008(From: HTUTUUUTTH)1、PCR产物测序时出现重叠峰问题图1(模板中有碱基缺失,往往是单一位点(1-1)或两个位点(1-2)碱基缺失导致测序结果移码)图1-1图1-2解决方法:将PCR产物克隆到质粒(如T载体)中挑单克隆测序,或将PCR 产物进行PAGE纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序。
问题图2(PCR产物不纯,含部分序列一致的两种以上的片段,长度不一)图2解决方法:主要原因是PCR产物没有纯化,含有部分序列一致的两种以上长度不一的片段,将PCR产物进行PAGE纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序,便可解决。
问题图3(测序引物有碱基缺失)测序引物有碱基缺失(一般是引物的5'端缺失),和模板的碱基缺失即图1有些类似,所不同的是模板碱基缺失一般是在一段正常测序序列后才出现移码,而引物碱基缺失的话,则从测序一开始就出现移码,表面在图形上便是一开始就是严重的峰形重叠。
解决方法:重新合成引物,或将引物进行PAGE纯化2、克隆测序时出现峰形重叠原因:所挑选的重组子不是单克隆,所提供的测序用质粒中含有两种以上插入片段不同的质粒;或是是送测序的菌液污染解决方法:重新挑单克隆的菌落(划线分离单菌落),提质粒或送菌液再次测序。
3、样品有杂合/突变位点模板中有杂合型突变,也就说模板本身在这个位点出现突变;或者是从基因组中扩增出来的杂合位点。
如果模板有杂合(突变或缺失),那么测序图形中其他的位點一般都是單一的峰形,然后突然在某一個位點出現重叠峰(如图中箭頭所示)。
解决方法:建议将DNA片段克隆到载体再测序。
4、poly A/T和C/G cluster导致的套峰和测序信号衰减图4-1图4-3图4-4RACE测序时经常遇到图4-1和图4-4的情形,解决方法:从另一端测序;但如果这样的序列出现在中间,呵呵,目前还没有很好的解决方法,要看测序公司的本事了。

DNA测序常见问题分析及解决办法总结PCR类型测序模板注意事项PCR类型测序模板注意事项•总反应体积建议为50uL或100uL,扩增结束后取3ul用1%左右的琼脂糖电泳检测,应为单一的条带。
3ul样品总量不低于50ng(非常小的PCR产物可以酌情减小),PCR产物产量过低说明PCR扩增结果不是很理想,应改进条件重新扩增。
•对于有杂带和扩增弥散的PCR产物,需经琼脂糖电泳将目的片段切下来并回收,建议将PCR 产物作克隆后进行测序。
有杂带的和扩增弥散的PCR产物即使通过琼脂糖电泳纯化,测序仍有可能出现双峰等异常结果。
•经上述检测合格的PCR产物,需经过琼脂糖电泳纯化去除未反应的引物,dNTP,引物二聚体等影响测序反应的组分。
有多种纯化方法可供选择,Promega,Qiagen和生工等公司都有相应的产品可供选择。
纯化后的PCR产物经电泳检测估计总量应不低于200ng/Kb(非常小的PCR产物可以酌情减小)。
•与质粒模板相比,PCR产物彼此间差异很大,因此,每个PCR模板应尽可能提供相应的PCR 退火温度和PCR产物的长度,以供测序时参考。
•PCR模板一般不应短于200bp,过短的PCR产物应经克隆后进行测序。
•纯化好的PCR产物应溶于双蒸水中,TE缓冲液会严重影响测序反应。
•若让公司对PCR产物进行纯化,PCR产物需满足∶总量不低于1ug/kb,片段长度不低于200bp •各种PCR测序模板均应提供相应的测序引物,并尽可能提供引物的全序列,并将引物浓度准确稀释到5pmole/ul 。
引物浓度的换算关系∶总ng数 = pmole x 分子量/1000由于PCR产物测序相对较难,为使您能拿到一个好的结果,请尽量满足上述要求。
DNA测序常见问题分析及解决办法总结测序常见问题分析序列中出现N值的常见原因:通常有以下几种情况将造成测序结果中N值较多:•PCR产物直接进行测序,在PCR产物长度以后将无反应信号,机器将产生许多N值。

DNA测序技术的使用中常见问题DNA测序技术是一项重要的科学工具,被广泛应用于生物医学研究、进化学、司法科学等领域。
然而,在使用DNA测序技术的过程中,常常会遇到一些问题。
本文将介绍DNA测序技术使用中常见的问题,并提供相应的解决方案。
1. 样品质量问题在DNA测序之前,样品质量是至关重要的。
低质量的样品会导致测序结果的偏差和错误。
常见的样品质量问题包括样品的纯度不高、浓度不足或者样品受到了污染。
解决方案:- 提高样品纯化方法:使用恰当的纯化试剂和技术来提高样品的纯度,例如使用芳香族化合物或异维酮类物质。
- 检测样品浓度:使用光谱法或者比色法来测量样品中DNA的浓度,确保其符合测序要求。
- 避免样品污染:在样品处理过程中,严格遵守无菌操作规程,使用经过无菌过滤器过滤的试剂和器具。
2. 序列读取长度问题DNA测序技术可以产生从数十个碱基对到上百万个碱基对的序列。
然而,部分DNA测序技术的读取长度有限,这可能影响到某些研究或应用的结果。
解决方案:- 选择合适的测序方法:不同的测序方法具有不同的读取长度,选择适合自己研究目的的测序方法,以满足相关的研究需求。
- 使用测序技术的新发展:不断发展的测序技术,如第三代测序技术,具有较长的读取长度和更高的准确性,可以解决传统测序方法的限制。
3. 突变检测问题DNA测序技术不仅可以用于确定特定序列的顺序,还可以用于检测和鉴定突变。
然而,突变的检测可能受到多种因素的影响,如检测方法的灵敏度和特异性、突变类型的多样性等。
解决方案:- 选择合适的突变检测方法:不同的突变具有不同的检测方法,如SNP分析、插入和缺失突变的分析等。
选择合适的方法来检测特定类型的突变。
- 结合多种检测方法:结合使用不同的突变检测方法可以提高突变检测的准确性和可靠性。
- 验证突变结果:通过使用其他独立的测序技术或方法来验证突变结果,以确保结果的可靠性。
4. 数据分析问题DNA测序技术生成的数据量巨大,数据分析是一个复杂而耗时的过程。

1 、为什么开始一段序列的信号很杂乱,几乎难以辨别?这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生N 值,峰图较杂?由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列?如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点?可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事? 首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)?是否会影响测序结果?序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
7 、测序结果为什么与标准序列有差别?原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。

1 、为什么开始一段序列的信号很杂乱,几乎难以辨别这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生 N 值,峰图较杂由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生 N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)是否会影响测序结果序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
7 、测序结果为什么与标准序列有差别原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。


1 、为什么开始一段序列的信号很杂乱,几乎难以辨别?这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生N 值,峰图较杂?由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列?如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点?可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事? 首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)?是否会影响测序结果?序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
7 、测序结果为什么与标准序列有差别?原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。

D N A测序结果中常见的几个问题公司内部档案编码:[OPPTR-OPPT28-OPPTL98-OPPNN08]1 、为什么开始一段序列的信号很杂乱,几乎难以辨别这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生 N 值,峰图较杂由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)是否会影响测序结果序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
DNA测序过程可能遇到的问题及分析对于一些生物测序公司(如Invitrogen等),我们的菌液或质粒经过PCR和酶切鉴定都没问题,但几天后的测序结果却无法另人满意。
为什么呢?PCR产物直接进行测序,在PCR产物长度以后将无反应信号,机器将产生许多N值。
这是由于Taq酶能够在PCR反应的末端非特异性地加上一个A碱基,我们所用的T载体克隆PCR产物就是应用该原理,通常PCR产物结束的位点,PCR产物测序一般末端的一个碱基为A(绿峰),也就是双脱氧核甘酸ddNTP终止反应的位置之前的A,A后的信号会迅速减弱。
N值情况一般是由于有未去除的染料单体造成的干扰峰。
该干扰峰和正常序列峰重叠在一起,有时机器377以下的测序仪无法正确判断出为何碱基。
有时,在序列的起始端的小片段容易丢失,导致起始区信号过低,机器有时也无法正确判读。
在序列的3’端易产生N值。
一个测序反应一般可以读出900bp以上的碱基(ABI3730可以达到1200bp),但是,只有一般600bp以前的碱基是可靠的,理想条件下,多至700bp的碱基都是可以用的。
一般在650bp以后的序列,由于测序毛细管胶的分辩率问题,会有许多碱基分不开,就会产生N值。
测序模板本身含杂合序列,该情况主要发生在PCR产物直接测序,由于PCR产物本身有突变或含等位基因,会造成在某些位置上有重叠峰,产生N值。
这种情况很容易判断,那就是整个序列信号都非常好,只有在个别位置有明显的重叠峰,视杂合度不同N值也不同。
测序列是从引物3’末端后第一个碱基开始的,所以就看不到引物序列。
有两种方法可以得到引物序列。
1.对于较短的PCR产物(<600bp),可以用另一端的引物进行测序,从另一端测序可以一直测通,可以在序列的末端得到该引物的反向互补序列。
对于较长的序列,一个测序反应测不通,就只能将PCR产物片段克隆到载体中,用载体上的通用引物(T7/SP6)进行测序。
载体上的通用引物与所插入序列间有一段距离,因此就可以得到完整的引物序列。

测序常见问题分析与解答1、DNA测序样品用什么溶液溶解比较好?答:溶解DNA测序样品时,用灭菌蒸馏水溶解最好。
DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件。
如果DNA用缓冲液溶解后,在进行了测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。
有很多客户在溶解DNA测序样品时使用TE Buffer。
的确,TE Buffer能增加DNA样品保存期间的稳定性,但TE Buffer对DNA测序反应有影响,根据我们的经验,我们还是推荐使用灭菌蒸馏水来溶解DNA测序样品。
2、提供DNA测序样品时,提供何种形态的比较好?答:我们推荐客户提供菌体,由我们来提取质粒,这样DNA样品比较稳定。
如果您要以提供DNA样品,我们也很欢迎,但一定要注意样品纯度和数量。
提供的测序样品为PCR产物时,特别需要注意DNA的纯度和数量。
PCR产物应该进行切胶回收,否则无法得到良好的测序效果。
有关DNA测序样品的详细情况请严格参照“DNA测序样品准备及注意事项”部分的说明。
3、提供的测序样品为菌体时,以什么形态提供为好?答:一般菌体的形态有:平板培养菌、穿刺培养菌,甘油保存菌或新鲜菌液等。
我们提倡寄送穿刺培养菌或新鲜菌液。
平板培养菌运送特别不方便,我们收到的一些平板培养菌的培养皿在运送过程中常常已经破碎,面目全非,需要用户重新寄样。
这样既误时间,又浪费客户的样品。
一旦是客户非常重要的样品时,其后果更不可设想。
而甘油保存菌则容易污染。
制作穿刺菌时,可在1。
5ml的Tube管中加入琼脂培养基,把菌体用牙签穿刺于琼脂培养基(固体)中,37℃培养一个晚上后便可使用。
穿刺培养菌在4℃下可保存数个月,并且不容易污染,便于运送。
4、与测序引物有关的问题:答:对于通用测序引物,只要正确使用,一般不会有太大问题,测序引物问题主要发生在客户自己提供的PCR引物上。
应该明确的一点是并不是所用的用于PCR的引物都可以用来作测序,以下几种PCR引物将是不适合用作测序引物的:(1)简并引物,简并引物必然要在测序模板上有多个结合位点,直接影响测序结果。

2 结果2.1 成功的建立HSV-1致死性感染动物模型,HSV感染滴度为106ID50/0.1ml。
死亡小鼠各组织器官涂片上HSV抗原均呈阳性反应,表明小鼠死于HSV感染。
2.2 空白对照组小鼠100%健康存活,致死攻击组小鼠100%发病死亡。
多于感染后第4~5天出现茸毛、抽搐、下肢麻痹等脑炎症状,5~7d内死亡。
M cAbS被动免疫实验组对小鼠均有保护作用。
第3、4组小鼠全部存活21d以上,第5组于感染后第15天发生1只鼠死亡,其余全部存活。
于感染后24d将第1,3,4,5组小鼠人工处死。
以上表明HSV-1 M cAbS对HSV-1致死性感染的小鼠几乎100%有特异性保护治疗作用。
另外,致死攻击组小鼠各组织器官涂片HSV 抗原均呈强阳性,其余各组与对照组为阴性。
表明小鼠感染HSV后,发生病毒血症导致死亡。
3 讨论HSV属于疱疹病毒科的具有代表性的典型疱疹病毒,其核酸为线状双股DNA。
有两种血清型:HSV-1和HSV-2。
两型病毒的DNA有50%的同源性,因此有型间共同抗原,也有型特异性抗原。
HSV病毒体有6种与细胞膜相关的糖蛋白,分别为糖蛋白B(g B)、gC、gD、g E、gG和g H。
其中gB、g D 与病毒感染的吸附及穿入有关,gH与病毒释放有关[7]。
g D 是HSV-1和HSV-2的共同抗原决定簇,其抗gD抗体属中和抗体,中和病毒的能力最强,能与HSV的g D抗原特异结合,结果改变了病毒的表面结构,阻断了病毒对宿主细胞的吸附,使病毒失去了感染能力[8]。
本实验所用的M cAbS中既有能识别HSV的gB、g D抗原的型共同性特异性单克隆抗体,也有能识别HSV的g C的型特异性单克隆抗体,体外中和试验也证明其中和效价为105~107,具有很强的中和病毒的作用[9]。
本实验结果在观察期内,空白对照组100%存活,且HSV抗原呈阴性。
致死对照组100%死亡,HSV抗原呈强阳性,证实本组小鼠死于HSV感染。
1 、为什么开始一段序列的信号很杂乱,几乎难以辨别?这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生N 值,峰图较杂?由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列?如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点?可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事? 首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)?是否会影响测序结果?序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
7 、测序结果为什么与标准序列有差别?原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。

DNA测序常见问题解析DNA测序常见问题解析⼀.引物问题Q:为什么我在测序报告上找不到我的引物序列?A:这⾥分以下⼏种情况:1. PCR引物作为测序引物进⾏测序时,所测序列是从引物3'末端后第⼀个碱基开始的,⽽且刚刚开始的碱基由于在⽑细管电泳中不能很好地分离⽽导致准确性下降,所以找不到您的引物序列。
(1)对于较短的PCR产物(<600 bp),可以⽤另⼀端的引物进⾏测序,从另⼀端测序可以⼀直测到序列的末端,就可以在序列的末端得到您的引物的反向互补序列。
(2)对于较长的序列,⼀个测序反应测不到头,因此就只能将您的PCR产物⽚段克隆到适当的载体中,⽤载体上的通⽤引物进⾏测序。
由于载体上的通⽤引物与您的插⼊序列之间还有⼀段距离,因此就可以得到您的完整的引物序列。
(3)由于在测序的起始端总会有⼀些碱基⽆法准确读出,因此,您如果想得到您的PCR产物的完整序列,最好克隆后进⾏测序。
2. 有时质粒做模板进⾏测序时,由于某些原因,质粒上没有插⼊外源⽚段,为空载体,所测的序列完全为载体序列,此时也找不到引物序列。
3. 找不到克隆⽚段的扩增引物。
发⽣这种情况原因有2个:(1)您在构建质粒时采⽤的⼯具酶的酶切位点距离您的测序引物太近,由于荧光染料的⼲扰在序列开始的部分会不⼗分准确。
⽐如pBluescript Ⅱ KS这个质粒如果采⽤Sac I做⼯具酶,采⽤T7引物测序:那么从T7引物末端到Sac I的酶切位点只有6个bp,这样酶切位点后的扩增引物序列在测序报告上很可能不完整。
解决的办法是采⽤M13 Forward引物来测序,这样可以保证Sac I的位点和之后的引物序列都可以完整的出现在报告中。
(2)您的插⼊⽚段的插⼊⽅向是反的,这时您不妨找⼀下您引物的反向互补序列。
或者您插⼊的⽚段可能不是您的⽬的⽚段,⽽是由于⾮特异性扩增出来的⽚段,还有可能您送过来的样品被污染。
Q:测序发现引物有突变或缺失是什么原因?A:测序发现引物区有突变,主要考虑三个⽅⾯的原因:测序,PCR/克隆过程,引物本⾝。

当然尽管我们有1 、 为什么开始一段序列的信号很杂乱,几乎难以辨别 这主要是因为残存的染料单体造成的干扰峰所致, 该干扰峰和正常序列峰重叠在一起; 另外, 测序电泳开始阶段电压有一个稳定期, 所以经常有 20-50 bp 的紧接着引物的片段读不清楚, 有时甚至更长。
2 、 为什么在序列的末端容易产生 N 值,峰图较杂 由于测序反应的信号是逐渐减弱的, 所以序列末端的信号会很弱, 峰图自然就会杂乱, 加上 测序胶的分辨率问题, 如果碱基分不开, 就会产生 N 值,正常情况下 ABI377 测序仪能正确 读出 500 个碱基的有效序列。
3 、 测序结果怎么找不到我的引物序列 如果找不到测序所用的引物序列。
这是正常的, 因为引物本身是不被标记的, 所以在测序报 告中是找不到的; 如果找不到克隆片段中的扩增引物, 可能是您克隆的酶切位点距离您的测 序引物太近,开始一段序列很杂,几乎难以辨别, 有可能看不清或看不到扩增引物;另外插 入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、 测序结果怎么看不到我克隆的酶切位点 可能的原因同上, 您克隆的酶切位点距离您的测序引物太近, 开始一段序列很杂, 几乎难以 辨别, 有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物, 当 然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、 你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事首先, 我们会核实给您的测序结果是否对应您的样品编号, 如果对应的是您的样品, 由于不 知您的实验背景, 测得的序列是否与您预想的结果一致我们无法判断, 我们能做到的是检查 发送给您的测序结果和您提供来的样品是否一致。
6 、 序列图为什么会有背景噪音 ( 杂带 ) 是否会影响测序结果 序列图的背景杂带是由荧光染料引起, 如果太强会影响测序结果, 要看信噪比, 我们给的结 果信噪比大都在 98%以上。

DNA测序结果分析比对 - 测序图初识通常一份测序结果图由红、黑、绿和蓝色测序峰组成,代表不同的碱基序列。
测序图的两端(本图原图的后半段被剪切掉了)大约50个碱基的测序图部分通常杂质的干扰较大,无法判读,这是正常现象。
这也提醒我们在做引物设计时,要避免将所研究的位点离PCR序列的两端太近(通常要大于50个碱基距离),以免测序后难以分析比对。
我的课题是研究基因多态性的,因此下面要介绍的内容也主要以判读测序图中的等位基因突变位点为主。
实际上,要在一份测序图中找到真正确实的等位基因多态位点并不是一件容易的事情。
由于临床专业的研究生,这些东西是没人带的,只好自己研究。
开始时大概的知道等位基因位点在假如在测序图上出现像套叠的两个峰,就是杂合子位点。
实际比对了数千份序列后才知道,情况并非那么简单,下面测序图中标出的两个套峰均不是杂合子位点,如图并说明如下:说明:第一组套峰,两峰的轴线并不在同一位置,左侧的T峰是干扰峰;第二组套峰,虽两峰轴线位置相同,但两峰的位置太靠近了,不是杂合子峰,蓝色的C峰是干扰峰通常的杂合子峰由一高一略低的两个轴线相同的峰组成,此处的序列被机器误判为“C”,实际的序列应为“A”,通常一个高大碱基峰的前面1~2个位点很容易产生一个相同碱基的干扰峰,峰的高度大约是高大碱基峰的1/2,离得越近受干扰越大。
一个摸索出来的规律是:主峰通常在干扰峰的右侧,干扰峰并不一定比主峰低。
最关键的一点是一定要拿疑似为杂合子峰的测序图位点与测序结果的文本序列和基因库中的比对结果相比较;一个位点的多个样本相比较;你得出的该位点的突变率与权威文献或数据库中的突变率相比较。
通常,对于一个疑似突变位点来说,即使是国际上权威组织大样本的测序结果中都没有报道的话,那么单纯通过测序结果就判定它是突变点,是并不严谨的,因一份PCR产物中各个碱基的实际含量并不相同,很难避免不产生误差的。
对于一个未知突变位点的发现,通常还需要用到更精确的酶切技术。

DNA测序技术改进方法建议随着近年来生物技术的飞速发展和普及,DNA测序技术已成为生物科学研究和医学诊断领域的重要工具。
然而,目前的DNA测序技术还存在一些挑战和局限,需要改进和完善。
在这篇文章中,我将针对当前DNA测序技术面临的问题,提出一些建议和改进方法。
首先,现有的DNA测序技术中,尽管高通量测序技术已经取得了巨大进展,但其测序错误率仍然较高。
因此,改进测序准确度是一个至关重要的问题。
为此,我们可以采取以下措施:1. 引入错误校正算法:通过在测序过程中引入错误校正算法,例如使用基于质量值和碱基频率的算法,可以有效降低测序错误率。
此外,结合深度学习技术,可以提高错误校正的准确性和效率。
2. 优化DNA库的构建:当前DNA测序技术中,DNA库的构建对测序准确度至关重要。
因此,我们可以通过优化DNA库构建的化学反应条件、选择更高质量的DNA样品和适当的文库制备方法等方式来提高测序准确度。
其次,高通量测序技术尽管可以快速获得大量的测序数据,但其对于长DNA片段的测序仍然存在困难。
因此,我们需要采取一些措施,以提高长DNA片段的测序质量和效率:1. 引入第三代测序技术:第三代测序技术具有快速、高效和长读长等优势,可以克服短读长引起的重叠区域难以拼接的问题。
因此,引入第三代测序技术作为补充,可以有效提高长DNA片段的测序质量。
2. 优化测序反应条件:目前,DNA测序中常使用的链终止法和桥式扩增法在长DNA片段的测序中存在一定的局限性。
因此,我们可以通过优化反应条件,例如优化引物和酶的浓度和反应温度等,来提高长DNA片段的测序效率。
此外,DNA测序技术的高昂成本也是制约其广泛应用的一个重要因素。
为了降低测序成本,我们可以采用以下措施:1. 提高测序效率:通过优化测序反应条件,例如缩短反应时间和提高反应效率,可以提高单位时间内的测序数据产量,从而降低测序成本。
2. 推广经济实惠的测序平台:当前市场上已经出现了一些经济实惠的DNA测序平台,如小型台式测序仪等。