An Exhaustive Genome Assembly Algorithm Using K-Mers to Indirectly Perform N-Squared Compar
- 格式:pdf
- 大小:66.10 KB
- 文档页数:2

高尔基体概述高尔基体(Golgi apparatus)是由许多扁平的囊泡构成的以分泌为主要功能的细胞器。
又称高尔基器或高尔基复合体;在高等植物细胞中称分散高尔基体。
最早发现于1855年,1898年由意大利人卡米洛•高尔基(Camillo Golgi,1844-1926)在光学显微镜下研究银盐浸染的猫头鹰神经细胞内观察到了清晰的结构,因此定名为高尔基体。
因为这种细胞器的折射率与细胞质基质很相近,所以在活细胞中不易看到。
高尔基体从发现至今已有100多年的历史,其中一半以上的时间是进行关于高尔基体的形态甚至是它是否真实存在的争论。
细胞学家赋予它几十种不同的名称,也有很多人认为高尔基体是由于固定和染色而产生的人工假像。
直到20世纪50年代应用电子显微镜才清晰地看出它的亚显微结构。
它不仅存在于动植物细胞中,而且也存在于原生动物和真菌细胞内。
形态与组成高尔基体是由数个扁平囊泡堆在一起形成的高度有极性的细胞器。
常分布于内质网与细胞膜之间,呈弓形或半球形,凸出的一面对着内质网称为形成面(forming face)或顺面(cis face)。
凹进的一面对着质膜称为成熟面(mature face)或反面(trans face)。
顺面和反面都有一些或大或小的运输小泡,在具有极性的细胞中,高尔基体常大量分布于分泌端的细胞质中。
顺面和反面都有一些或大或小的运输小泡(图6-24),在具有极性的细胞中,高尔基体常大量分布于分泌端的细胞质中(图6-25)。
图6-24高尔基体各部分的名称图6-25培养的上皮细胞中高尔基体的分布(高尔基体为红色,核为绿色)引自/因其看上极像滑面内质网,因此有科学家认为它是由滑面内质网进化而来的。
扁平囊的直径为1μm,由单层膜构成,膜厚6~7nm,中间形成囊腔,周缘多呈泡状,4~8个扁平囊在一起,某些藻类可达一二十个,构成高尔基体的主体,称为高尔基堆(Golgi stack)。
高尔基体膜含有大约60%的蛋白和40%的脂类,具有一些和ER共同的蛋白成分。

eubacterium分类-回复Eubacteria Classification: Exploring the Diversity of the Eubacteria KingdomIntroduction:The classification of living organisms helps scientists understand and organize the vast diversity of species on our planet. One such classification is the categorization of bacteria into different kingdoms. The Eubacteria kingdom, also known as true bacteria, encompasses a wide range of organisms, each with unique characteristics and ecological roles. In this article, we will delve into the classification of Eubacteria, exploring the various groups and their distinguishing features.Historical Background:The study of bacteria classification dates back to the early 17th century when the Dutch scientist Antonie van Leeuwenhoek observed bacteria under a microscope for the first time. However, it was not until the groundbreaking work of the German microbiologist Carl Woese in the 1970s that a better understandingof bacteria classification emerged. Woese proposed a system based on the sequencing of ribosomal RNA (rRNA) genes, which allowed for a more accurate classification of microorganisms.Classification of Eubacteria:The Eubacteria kingdom is divided into multiple phyla, each containing different classes, orders, families, genera, and species. While the exact number of phyla is subject to ongoing research and debate, some of the most well-known and extensively studied phyla include Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes.1. Proteobacteria:The Proteobacteria phylum comprises a diverse group of bacteria with varying shapes and metabolic capabilities. This phylum is subdivided into several classes, including Alpha-, Beta-, Gamma-, Delta-, and Epsilonproteobacteria. Alpha-proteobacteria include several symbiotic and pathogenic species like Rhizobium and Agrobacterium. Beta-proteobacteria often inhabit aquatic environments and include nitrifying bacteria such as Nitrosomonas.Gamma-proteobacteria include many clinically significant bacteria like Escherichia coli and Pseudomonas aeruginosa. Delta- and Epsilonproteobacteria encompass species capable of inhabiting extreme environments, such as deep-sea hydrothermal vents.2. Firmicutes:The Firmicutes phylum consists of bacteria with a Gram-positive cell wall structure. This phylum is further divided into several classes, including Bacilli and Clostridia. Bacilli include well-known pathogenic species like Staphylococcus and Streptococcus. Moreover, Bacillus subtilis, a species in this class, serves as a model organism for studying bacterial biofilms. Clostridia include notable species such as Clostridium botulinum, responsible for botulism, and Clostridium tetani, the causative agent of tetanus.3. Actinobacteria:The Actinobacteria phylum is known for its filamentous structure and includes many different classes, such as Actinobacteria, Acidimicrobiia, and Thermoleophilia. Actinobacteria, often referred to as Actinomycetes, include numerous species involved in theproduction of antibiotics, such as Streptomyces and Mycobacterium tuberculosis, the bacterium responsible for tuberculosis. Acidimicrobiia and Thermoleophilia comprise thermophilic or acidophilic species found in extreme environments.4. Bacteroidetes:The Bacteroidetes phylum consists of Gram-negative bacteria found in diverse habitats, including soil, water, and the guts of animals. This phylum is characterized by its ability to degrade complex carbohydrates. Notable genera within Bacteroidetes include Bacteroides and Prevotella, which play essential roles in the digestion process within the intestines of humans and animals.Conclusion:The classification of bacteria provides scientists with a systematic approach to identify and study the diverse array of species within the Eubacteria kingdom. This classification allows researchers to understand the ecological significance, evolutionary relationships, and potential applications of different bacterial groups. While the classifications discussed in this article are just a glimpse into thevast diversity present in the Eubacteria kingdom, they provide an important foundation for further exploration and understanding of these microscopic organisms.By delving into the classification of Eubacteria, we can gain a deeper appreciation for the intricate and diverse nature of these microorganisms. Advances in molecular techniques and ongoing research will undoubtedly contribute to future refinements in the classification system, providing us with an even more precise understanding of the bacterial world.。

pre-gastrulation developmentalWhat is Pre-gastrulation Developmental Phase?Pre-gastrulation developmental phase refers to the early stage in embryonic development before the formation of the gastrula. During this critical phase, various crucial events occur that lay the foundation for the subsequent formation of the three germ layers that give rise to the different tissues and organs in the developing embryo. In this article, we will explore the pre-gastrulation developmental phase in detail, discussing its key stages and the processes that take place during this time.1. Fertilization and Cleavage:The pre-gastrulation phase begins with fertilization, where a sperm fuses with an egg to form a zygote. Following fertilization, the zygote undergoes cleavage, a process of rapid cell divisions. These divisions result in the formation of blastomeres, smaller cells that make up the blastula.2. Blastula Formation:As cleavage continues, the blastomeres divide and rearrange, leading to the formation of a hollow ball-like structure called ablastula. The blastula consists of an outer layer of cells, known as the trophoblast, and an inner cell mass.3. Compaction and Morula Formation:During this stage, the blastomeres undergo a process called compaction, where they tightly adhere to each other, forming a compacted ball of cells called a morula. Compaction is crucial for the subsequent differentiation of embryonic cells.4. Blastocyst Formation:At this point, the morula undergoes further cell divisions and differentiation, resulting in the formation of a blastocyst. The blastocyst consists of two distinct cell populations: the inner cell mass (ICM) and the outer trophoblast layer. The ICM gives rise to the embryo, while the trophoblast layer contributes to the formation of extraembryonic structures such as the placenta.5. Implantation:The blastocyst moves towards the uterine lining and undergoes implantation, a process where it buries itself into the endometrium. This establishes a connection between the embryo and the maternal blood supply, allowing for nutrient and gas exchange.6. Formation of the Three Germ Layers:Following implantation, the pre-gastrulation phase progresses further as the blastocyst differentiates into the three germ layers: ectoderm, mesoderm, and endoderm. This process is known as gastrulation. The ectoderm gives rise to the nervous system, skin, and other ectodermal tissues. The mesoderm gives rise to the skeletal system, muscles, heart, and blood vessels. The endoderm gives rise to the gastrointestinal tract, respiratory system, and other endodermal tissues.7. Germ Layer Migration and Differentiation:During gastrulation, cells from each of the three germ layers undergo migration and differentiation to form specific tissues and organs. For example, ectodermal cells migrate to form the neural tube, which develops into the brain and spinal cord. Mesodermal cells differentiate to form muscles, bones, and internal organs. Endodermal cells give rise to the lining of the digestive and respiratory tracts.8. Organogenesis:As gastrulation progresses, the three germ layers continue todifferentiate and form the rudiments of various organs. This process, known as organogenesis, involves intricate cell interactions, proliferation, and remodeling to shape and develop organs such as the heart, lungs, liver, and kidneys.In conclusion, the pre-gastrulation developmental phase is a crucial period in embryonic development. It involves key events such as fertilization, cleavage, blastula, and blastocyst formation, implantation, gastrulation, and organogenesis. These processes play a fundamental role in establishing the basic body plan of the developing embryo, paving the way for its subsequent growth and differentiation into a complex multicellular organism.。

应用地球化学元素丰度数据手册迟清华鄢明才编著地质出版社·北京·1内容提要本书汇编了国内外不同研究者提出的火成岩、沉积岩、变质岩、土壤、水系沉积物、泛滥平原沉积物、浅海沉积物和大陆地壳的化学组成与元素丰度,同时列出了勘查地球化学和环境地球化学研究中常用的中国主要地球化学标准物质的标准值,所提供内容均为地球化学工作者所必须了解的各种重要地质介质的地球化学基础数据。
本书供从事地球化学、岩石学、勘查地球化学、生态环境与农业地球化学、地质样品分析测试、矿产勘查、基础地质等领域的研究者阅读,也可供地球科学其它领域的研究者使用。
图书在版编目(CIP)数据应用地球化学元素丰度数据手册/迟清华,鄢明才编著. -北京:地质出版社,2007.12ISBN 978-7-116-05536-0Ⅰ. 应… Ⅱ. ①迟…②鄢…Ⅲ. 地球化学丰度-化学元素-数据-手册Ⅳ. P595-62中国版本图书馆CIP数据核字(2007)第185917号责任编辑:王永奉陈军中责任校对:李玫出版发行:地质出版社社址邮编:北京市海淀区学院路31号,100083电话:(010)82324508(邮购部)网址:电子邮箱:zbs@传真:(010)82310759印刷:北京地大彩印厂开本:889mm×1194mm 1/16印张:10.25字数:260千字印数:1-3000册版次:2007年12月北京第1版•第1次印刷定价:28.00元书号:ISBN 978-7-116-05536-0(如对本书有建议或意见,敬请致电本社;如本社有印装问题,本社负责调换)2关于应用地球化学元素丰度数据手册(代序)地球化学元素丰度数据,即地壳五个圈内多种元素在各种介质、各种尺度内含量的统计数据。
它是应用地球化学研究解决资源与环境问题上重要的资料。
将这些数据资料汇编在一起将使研究人员节省不少查找文献的劳动与时间。
这本小册子就是按照这样的想法编汇的。

Development of an industrial medium and a novel fed-batch strategy for high-level expression of recombinant b -mananase by Pichia pastorisa a ,b a a a a a ,b ,⇑fermenter.a r t i c l e i n f o Article history:Received 2March 2012Received in revised form 13May 2012Accepted 14May 2012Available online 23May 2012Keywords:Pichia pastoris MannanaseFed-batch cultivation pGAPa b s t r a c tAn industrial medium,Corn Steep Liquor Powder Dextrose (CSD medium)was developed for constitutive expression of recombinant b -mananase by Pichia pastoris .The b -mananase activity (513U/mL)with CSD medium was 1.64-and 2.5-fold higher than with YPD and BSM in shaken flasks.The b -mananase produc-tivity with CSD medium was 61.0U/mL h,which was 1.7-and 2.5-fold higher than with YPD and BSM in a 5-L fermenter based on a novel fed-batch strategy combining the real-time exponential feed mode with the DO-stat feed mode.The b -mananase activity,dry cell weight and the recombinant enzyme reached up to 5132U/mL,110.0g/L and 4.50g/L after 50h cultivation in a 50-L fermenter.The high efficient expres-sion of recombinant b -mananase by P.pastoris indicated that CSD medium and the novel fed-batch strat-egy have great potential for the production of recombinant b -mananase in industrial fermentation.Ó2012Elsevier Ltd.All rights reserved.1.IntroductionMannans,widely distributed in wood,tubers,plant seeds,beans and cell walls of certain marine algae (Gübitz et al.,2001),are a major component of hemicelluloses (Petkowicz et al.,2001).Endo-1,4-b -D -mannanase (b -mannanase,EC 3.2.1.78)randomly hydrolyzes (1?4)-beta-D -mannosidic linkages in mannans,galac-tomannans and glucomannans (http://www.expasy.ch/enzyme/3.2.1.78).It has been applied in papermaking,food,animal feed,drilling industries and the production of second generation biofu-els (Dhawan and Kaur,2007;Moreira and Filho,2008),and many mannanase genes have been cloned and expressed in Pichia pasto-ris (Bien-Cuong et al.,2009;Luo et al.,2009).The methylotrophic yeast P.pastoris has been widely used for the heterologous protein expression.In particular,the high-level expression of heterologous protein by P.pastoris has been obtained using pAOX1expression system (Luo et al.,2009;Schenk et al.,2007).However,the fermentation process in the AOX1-based expression system is difficult to control because the excessive accu-mulation of methanol inhibits the cell growth and reducesthe total yield of heterologous protein (Pal et al.,2006).Furthermore,there is a fire hazard using methanol as an inducer and it is inappropriate for the production of foods and drugs (Cereghino and Cregg,1999).Recently,some researchers employed constitutive glyceral-dehyde-3-phosphate dehydrogenase (GAP)promoter for heterolo-gous protein expression in P.pastoris and it also led to the high-level expression of target proteins (Goodrick et al.,2001;0960-8524/$-see front matter Ó2012Elsevier Ltd.All rights reserved./10.1016/j.biortech.2012.05.065⇑Corresponding author at:School of Minerals Processing and Bioengineering,Central South University,South Lushan Road 932,Changsha,Hunan,People’s Republic of China.Tel.:+8673188877216;fax:+8673188710804.E-mail address:zhouhb@ (H.Zhou).Li et al.,2010).Different from pAOX1expression system in which the expression of heterologous protein was induced by methanol, recombinant protein was constitutively expressed by P.pastoris in pGAP expression system.Thus,the pGAP expression system is more suitable for the industrial fermentation due to the elimination of methanol in fed-batch cultivation(Zhang et al.,2009).However, there are few investigations on the high density fermentation of P.pastoris using the pGAP expression system,especially in indus-trial applications.In P.pastoris expression system,proper fermentation conditions (carbon source,nitrogen source,dissolved oxygen and pH,etc.)are very important for heterologous protein expression.Culture med-ium is one key factor for the high-level expression of heterologous protein in P.pastoris.Yeast Extract Peptone Dextrose Medium (YPD),Basal Salts Medium(BSM)and Buffered Methanol-complex Medium(BMMY)were classical culture media in P.pastoris expres-sion system published by Invitrogen Corporation(Carlsbad,CA, USA)(Macauley-Patrick et al.,2005).However,YPD contains expensive yeast extract and peptone,while BSM and BMMY are not only expensive but also not convenient to use because of their complex components.Developing an effective and cheap industrial medium would overcome limitations for industrial applications, especially for the production of low value-added enzymes using pGAP expression system.In our previous study,a strain of recombinant mannan endo-1,4-b-mannosidase P.pastoris was constructed and the recombi-nant b-mannanase with an excellent property displayed potential applications in food,animal feed and the production of biofuels (Zhao et al.,2011).In this study,a novel industrial medium with low cost and simple components and a novel fed-batch strategy would be developed for the high-level expression of b-mannanase by P.pastoris using the pGAP expression system in5-L and50-L fermenters.2.Methods2.1.Strain and mediumIn our previous study,the recombinant mannan endo-1,4-b-mannosidase P.pastoris was constructed using the pGAPexpression system(Invitrogen)(Zhao et al.,2011).Yeast extract,peptone,glu-cose,corn steep liquor powder,fish meal,soybean meal,peanut cake powder,various salts,etc.were of industrial or food grade.2.2.Determination of the optimal nitrogen source and carbon-to-nitrogen ratioThe nitrogen contents of various nitrogen sources were deter-mined using Kjeldahl method(Kirk,1950).To determine the optimal nitrogen source,50mL culture med-ium containing4%glucose and the corresponding nitrogen source with carbon-to-nitrogen ratios of1.0,2.5,4.0or6.0were added to 250mL shakenflasks.The strain was inoculated into5mL YPD (10g/L yeast extract,20g/L peptone and20g/L glucose)and cul-tured for24h at28°C in50mL shakerflasks(250rpm)as seed. Then5mL seed culture was transferred to these media and cul-tured at28°C(250rpm).The supernatant was sampled every 12h for cell concentration measurement and enzyme activity assay.2.3.Optimization of cultivation conditions in shakenflasksEffects of inoculum volume,culture pH and glucose concentra-tion on b-mannanase production and cell growth were studied in shakenflasks using Corn Steep Liquor Powder Dextrose medium (CSD medium).To determine the effect of inoculum volume on b-mannanase production and cell growth,0.5,2.5or5mL seed culture(about 4.0Â108cells/mL)were transferred to50mL CSD medium(27g/ L corn steep liquor powder and20g/L glucose)and cultured at 28°C(250rpm)in250mL shakenflasks.To determine the effect of culture pH on b-mannanase produc-tion and cell growth,5mL seed culture was transferred to50mL CSD medium at pH4.0,5.0,6.0maintained by0.05M phosphate buffer and at natural pH(an initial pH of5.0),and cultured at 28°C(250rpm)in250mL shakenflasks.To determine the effect of the glucose concentration on b-man-nanase production and cell growth,5mL seed culture was trans-ferred to50mL CSD medium containing20,40or60g/L glucose with the carbon-to-nitrogen ratio of 6.0and cultured at28°C (250rpm)in250mL shakenflasks.The supernatant was sampled every12h for b-mannanase activity assay,pH value,cell and glucose concentration measurement.2.4.Fed-batch cultivation in5-L fermenter using YPD,BSM and CSD mediaThe fed-batch fermentation in a5-L fermenter(KF-5l,koBIO Tech.Co.,Ltd,Korea)was implemented with1.2L initial medium and 2.0L feeding volume using YPD medium(20g/L glucose, 10g/L yeast extract,20g/L peptone),BSM medium(20g/L glu-cose,26.7mL85%H3PO4,4.13g/L KOH,4g/L(NH4)2ÁSO4,0.38g/ L CaCl2,18.2g/L K2SO4,14.9g/L MgSO4Á2H2O,0.93g/L Ca-SO4Á2H2O,4.0mL PTM1)and CSD medium(20g/L glucose,27g/ L corn steep liquor powder).PTM1trace salts solution contained: 6.0g/L CuSO4Á5H2O,0.088g/L KI,3.0g/L MnSO4ÁH2O,0.2g/L Na2-MoO4Á2H2O,0.02g/L H3BO3,0.5g/L CoCl2Á6H20,20.0g/L ZnCl2, 65.0g/L FeSO4Á7H2O,0.2g/L Biotin,5.0mL/L H2SO4.The propor-tion of inoculation was10%(v/v)of the initial medium volume. 500g/L glucose(containing5g/L yeast extract and10g/L peptone for YPD medium,12.0mL PTM1for BSM medium,and10g/L corn steep liquor powder for CSD medium)was fed according to the equation F=0.081ÁX0ÁV0Áe l t in the previous study(Zhao et al., 2007),where X0and V0standed for the initial cell concentration and the initial medium volume,and initial l was determined according to the formula:l=ln2/t d,where t d referred to the bio-mass doubling time.The supernatant was sampled every1h for the determination of cell and glucose concentration.When glu-cose concentration was more than10g/L or below2g/L,the glu-cose feed rate was adjusted according to the equation F=0.081ÁX tÁV tÁe l t(t0Àt),where X t and V t standed for the cell con-centration and medium volume after cultivation for t h when glu-cose concentration was not over the range of2–10g/L,and t0 referred to the fermentation time from the beginning.The initial culture pH was5.0and pH value was gradually adjusted to6.0–6.5using25%(v/v)ammonia after it began to feed500g/L glu-cose.Dissolved oxygen(DO)level was controlled above20%by adjusting tank pressure,stirring speed and aeration.When tank pressure,stirring speed and aeration exceeded the maximum set values,500g/L glucose was fed with a constant feed rate to ensure that DO level was not less than20%.The supernatant was sampled every1or2h for the determination of b-mannanase activity,cell concentration or protease activity.2.5.Fed-batch cultivation in a50-L fermenter using CSD mediumThe fed-batch cultivation in a50-L fermenter(B.Braun BIOSTAT C plus)was implemented with18L initial medium and20L feed-ing volume using CSD medium as the above description.The258J.Zheng et al./Bioresource Technology118(2012)257–264proportion of inoculation was10%(v/v)of the initial medium vol-ume.The seed culture was obtained by the cultivation of the re-combinant mannan endo-1,4-b-mannosidase P.pastoris for14–18h in a5-L fermenter.Five hundred grams per liter of glucose was fed according to the equation F=0.081ÁX0ÁV0Áe l t as de-scribed in the above.The supernatant was sampled every4h for the determination of cell and glucose concentrations.When glu-cose concentration was more than10g/L or below2g/L,the glu-cose feed rate was adjusted according to the equation F=0.081ÁX tÁV tÁe l t(t0Àt)as described in the above.The initial cul-ture pH was5.0and pH value was gradually adjusted to6.0–6.5 using25%(v/v)ammonia when it began to feed500g/L gucose. DO level was controlled above40%by adjusting tank pressure,stir-ring speed and aeration.When dry cell weight reached to94.9g/L (500g/L wet cell weight),500g/L glucose was fed with a constant fed rate to ensure that DO level was not less than40%.The super-natant was sampled every4h for the determination of b-mannan-ase activity and biomass.2.6.Cell concentration measurementDry cell weight(WCW)was determined by centrifugation(Cen-trifuge AG22331,Eppendorf,Hamburg,Germany)at10,000g for 5min and drying to a constant weight at80°C.2.7.The estimation of the amount of recombinant mannan endo-1,4-b-mannosidaseThe amount of recombinant mannan endo-1,4-b-mannosidase (Man26A)was estimated by using the correlation between the b-mannanase activity(U)and the protein mass(mg):1mg-Man26A=1139.2U,according to our previous study(Zhao et al., 2011).2.8.Mannan endo-1,4-b-mannosidase assayMannan endo-1,4-b-mannosidase activity was determined using the3,5-dinitrosalicylic acid(DNS)method as described in our previous study(Zhao et al.,2011).One unit of endo-1,4-b-man-nanase activity was defined as the amount of enzyme releasing 1l mol of mannose equivalents per minute.All experiments were done in triplicate.2.9.Protease assayFive milliliters of culture was centrifuged at10,000g for5min and supernatant was collected for protease activity assay accord-ing to Wu et al.’s method(Wu et al.,2008).2.10.Elemental analysis of corn steep liquor powder using ICPContents of metal and non-metallic elements in corn steep li-quor powder were determined using inductively coupled plasma optical emission spectrometry(ICP OES)according to the method of(Naozuka et al.(2011)).3.Results and discussion3.1.Development of CSD medium for the high-level expression of recombinant b-mananase by P.pastorisTo determine the optimal industrial medium for the high-level expression of recombinant b-mananase by P.pastoris,several indus-trial nitrogen sources,yeast extract and peptone were chosen as the sole nitrogen sources for cell growth of P.pastoris,respectively.Nitrogen contents determined by Kjeldahl method were12.75%for yeast extract and peptone(w/w,1/2),4.50%for corn steep liquor powder,8.00%forfish meal,5.60%for soybean meal and6.50%for peanut cake powder,respectively.Glucose was used as carbon source in these media.The optimal medium for the expression of re-combinant b-mananase was CSD medium,and afinal activity of 513.3U/mL was obtained,which is1.64-fold higher than with YPD medium(312U/mL)and3.2-fold higher than with BSM medium (156U/mL)(Table1).The dry cell weight was8.84g/L with CSD medium,which is1.7-fold higher than with YPD medium(5.30g/ L)and3.3-fold higher than with BSM medium(2.70g/L).The car-bon-to-nitrogen ratio had an very important effect on cell growth and protein expression of P.pastoris.Table1showed that the optimal carbon-to-nitrogen ratio was different when different nitrogen sources were used for recombinant b-mananase expression by P. pastoris.The optimal carbon-to-nitrogen ratios for cell growth and protein expression were2.5using yeast extract and peptone,4.0 using corn steep liquor powder and no more than2.5using peanut cake powder,fish meal and soybean meal.This may be due to that they were different in content of other ingredients such as amino nitrogen,vitamins,biotin and minerals.Elemental analysis of corn steep liquor powder using ICP showed that corn steep liquor powder was rich in phosphorus(10.300%)and magnesium(5.208%).Other mineral elements,Mn,Fe,Zn,K,Gu,As and Se were also detected, and their contents were0.047%,0.583%,0.144%,0.3575%,0.008%, 0.0005%and0.0002%,respectively.Corn steep liquor powder con-taining these compositions could provide plentiful nutrients for cell growth of P.pastoris.High b-mananase activity and biomass could be obtained over the carbon-to-nitrogen ratio of2.5–6.0with corn steep liquor powder as the nitrogen source.In P.pastoris expression system,yeast extract,peptone and ammonium salt were commonly used as nitrogen sources for the heterologous protein expression (Macauley-Patrick et al.,2005).This result revealed corn steep liquor powder was more suitable for recombinant b-mananase expression and P.pastoris growth than yeast extract,peptone and ammonium sulfate.And CSD medium(40g/L glucose and54g/L corn steep li-quor powder)could replace YPD and BSM media for the constitutive expression of recombinant b-mananase in P.pastoris with a higher expression level.3.2.Optimization of fermentation conditions in shakenflasksTo facilitate the study of fed-batch fermentation,the effects of inoculum,glucose concentration and pH value on the expression of recombinant b-mananase were investigated in shakenflasks. The results showed that inoculum volume had no significant ef-fect on b-mananase production andfinal cell concentration.How-ever,10%inoculum made cell enter into the logarithmic growth phase earlier.Thus,10%inoculum was taken up in the following fed-batch fermentation.60g/L glucose led to higher cell concen-tration and b-mananase activity,and20g/L glucose made cell enter into the decline phase earlier.When glucose concentration were20,40and60g/L,b-mananase yields per gram of glucose were17.1,13.6and10.1mg which were estimated by using the correlation between the b-mannanase activity(U)and the protein mass(mg)as described in materials and methods,and biomass yields(DCW)per gram of glucose were0.26,0.21and 0.18g,respectively.This indicated that20g/L of glucose resulted in a more efficient fermentation.Therefore,in fed-batch fermentation,glucose concentration in initial medium was20g/ L and it was controlled at a low level in the process of feeding. pH value also had an important effect on cell growth and heterologous protein expression by P.pastoris.It was reported that P.pastoris could grow well over pH range of3.0–7.0,and the optimal pH for heterologous protein expression was different for different proteins(Macauley-Patrick et al.,2005).Fig.1AJ.Zheng et al./Bioresource Technology118(2012)257–264259showed that the maximal b-mananase activity reached up to 341.9U/mL at uncontrolled pH,which was3.5-,1.2-,and1.1-fold higher than at pH4.0,5.0and6.0in0.05M phosphate buffer. Although the maximal cell densities at different pH values were basically the same,cells grew faster at uncontrolled pH than at pH4.0,5.0and6.0in0.05M phosphate buffer(Fig.1B).This also could be confirmed in Fig.1C,which showed that glucose was consumed faster at uncontrolled pH than at controlled pH by phosphate buffer.After cultivation for24h at uncontrolled pH, pH fell to the minimum pH value(pH4.4),and rose to6.5at 48h(Fig.1D).The results revealed that uncontrolled pH with a initial pH of5.0was more suitable for the expression of b-man-anase and cell growth.To further investigate the expression of recombinant b-manan-ase by P.pastoris,cultivation was performed in250mL shaken flasks using CSD medium at the optimal condition based on theTable1Mannan endo-1,4-b-mannosidase activity and biomass using various media with different carbon-to-nitrogen ratios.Various media The carbon-to-nitrogen ratio a Biomass(DCW b)(g/L)Activity(U/mL)YPD1 3.84±0.19240.2±0.32.5 5.30±0.12315.3±0.24 4.32±0.25270.1±0.46 2.37±0.37141.6±0.6PCD 2.5 5.83±1.00156.3±1.64 4.73±0.56126.3±0.96 2.96±0.31120.1±0.5FMD 2.5 5.82±1.06180.5±1.74 5.41±0.75148.0±1.2pH on b-mannanase production and cell growth.Time course of fermentation activity(A),cell growth(B),residualflasks containing50mL CSD medium at pH4.0(j),5.0(d),6.0(N)maintained by0.05M phosphate buffer and uncontrolled260J.Zheng et al./Bioresource Technology118(2012)257–264above results.The recombinant b -mananase activity rose with the increase in cell (Fig.2),which confirmed that the expression of re-combinant b -mananase was associated with cell growth (Zhang et al.,2009).After cell entered into the logarithmic phase,glucose concentration declined sharply,and pH value also declined,indi-cating that cell growth consumed large amounts of glucose and might lead to production of organic acids.After cultivation for 24h,pH value even fell to 4.1and began to rise,which might be explained that cell consumed a lot of organic nitrogen due to the lack of organic carbon (Hahn-Hägerdal et al.,2005).Cell concentra-tion and activity declined after cultivation for 60and 72h,respec-tively.The highest b -mananase activity,dry cell weight and the recombinant protein in supernatant were about 521.6U/mL,8.94g/L and 0.458g/L.3.3.Development of a novel fed-batch strategy in 5-L and 50-L fermenters using CSD mediumAccording to the above analysis,CSD medium (20g/L glucose,27g/L corn steep liquor powder)was used as the medium for the fed-batch cultivation of the recombinant P.pastoris in 5-L and 50-L fermenters as described in materials and methods.Oxygen and glu-cose were key factors for cell growth and protein expression (Lee et al.,2003).To further investigate the effects of glucose concentra-tion and dissolved oxygen (DO)on protein expression and cell growth,low DO level and glucose concentration stimulations were implemented when cell dry weight reached up to 19.0g/L (100g/L wet cell weight).With adequate glucose concentration,when DO le-vel was adjusted to 0–10%,cell concentration and b -mananase activity began to decrease.And alcohol was detected in culture.This revealed that P.pastoris might turn to anaerobic metabolic pathways and produce large amounts of ethanol under hypoxic conditionscarbon source led to the inhibition of cell growth,the increase of cost and a waste of resources (Tang et al.,2009).The results in shaken flasks also indicated that 20g/L glucose led to a higher b -mananase and biomass yields than 40and 60g/L glucose.To overcome these shortcomings,glucose was fed with the exponential feed mode and the feed rate was corrected in real time according to the feed-back results of glucose concentration in culture.At the same time,DO level was maintained over 20%by adjusting stir speed,ventila-tion and tank pressure.This ensured that cell could grow fastly and no excessive glucose was accumulated in culture.Based on the above findings,a novel fed-batch control strategy combining the real-time exponential feed mode and DO-stat feed mode was adopted to ensure the proper glucose concentration and DO level during the fed-batch process in a 5-L fermenter.As shown in Fig.3,the fed-batch cultivation could be divided into three stages:batch phase,exponential feeding phase and constant feeding phase.In the batch phase,uncontrolled pH with the ini-tial pH 5.0was carried out.The results in shaken flasks revealed that cultivation below pH 4.0resulted in a low b -mananase activ-ity and slow cell growth.Therefore,when pH was below 4.0,it was adjusted by feeding 25%(v/v)ammonia.After cultivation for 10h,glucose concentration decreased below 10g/L,and 500g/L glucose was fed as described in materials and methods.This led to a fast decrease of pH because cell grew fastly with the quick consumption of glucose and oxygen (Fig.3).According to the results in shaken flasks,pH value was adjusted to 6.0–6.5gradually by feeding ammonia in order to obtain a high level expression of b -mananase.After cultivation for 30h,due to high glucose feed rate,even the maximum tank pressure,stirring speed and aeration could’t maintain a high dissolved oxygen.Thus,DO-stat strategy was taken up to keep DO level above 20%by feeding glucose with a constant rate.In the constant feed-J.Zheng et al./Bioresource Technology 118(2012)257–264261in the 50-L fermenter which could provide higher stirring speeds,greater ventilation and higher tank pressure.Thus,during the fed-batch cultivation in the 50-L fermenter,DO level was main-tained above 40%.Based on this fed-batch strategy,the highest bio-mass was 110.0g/L (DCW)after 32h cultivation,which was 1.9-,2.2-and 2.1-fold higher than with YPD,BSM and CSD medium in a 5-L fermenter (Table 2).The highest b -mananase activity was 5132U/mL after 50h cultivation which was 2.34-, 2.53-and 2.33-fold higher than with YPD,BSM and CSD medium in a 5-L fer-menter (Table 2).The b -mananase activity in 50-L fermenter using CSD medium based on the novel fed-batch strategy was 16.3-,32.7-and 10-fold higher than with YPD,BSM and CSD media in 250mL shaken flasks.The recombinant enzyme in supernatant was 4.50g/L,which estimated by using the correlation between the b -mannanase activity (U)and the protein mass (mg)as de-scribed in materials and methods.The expression level using CSD medium in fermenters was significantly higher than that of any other recombinant b -mananase expressed by P.pastoris from Aspergillus sulphureus (96U/mL),Aspergillus niger BK01(669U/mL),Bispora sp.MEY-1(500U/mL in a 3.7-L fermenter)and Bacillus subtilis MA139(230U/mL)(Bien-Cuong et al.,2009;Chen et al.,2007;Luo et al.,2009;Qiao et al.,2010).parison of high-density fermentation of the recombinant P.pastoris using CSD,YPD and BSM mediaTo further evaluate CSD medium,fed-batch cultivation of the recombinant P.pastoris was implemented with CSD,YPD and BSM media based on the novel fed-batch strategy.The b -mananase activity and maximum dry cell weight reached up to 2029.2U/mL and 49.9g/L for BSM medium after 81h cultivation,2188.8U/mLand 58.0g/L for YPD medium after 57h cultivation,and 2195.5U/mL and 53.5g/L for CSD medium after 36h cultivation,respectively (Table 2).b -mananase productivity for BSM,YPD and CSD medium were 24.4,35.9and 61.0U/mL h,-pared with YPD and BSM media,the fermentation period was shortened from 81and 61h to 36h with CSD medium.The stagna-tion phase of cell growth with CSD medium was shorter than with YPD and BSM media.The overall specific growth rate of the recom-binant P.pastoris in BSM,YPD and CSD media were 0.028,0.046and 0.078h À1,respectively (Table 2).The results indicated that CSD medium resulted in more efficient protein expression and fas-ter cell growth than BSM and YPD media,which could reduce costs in power and labor.Generally,in lab scale and pilot scale fermen-tation,BSM was used for the heterologous protein expression with ammonium salts as the nitrogen source (Kui et al.,2010;Zhao et al.,2007).However,it was complex,expensive and difficult to prepare.It was reported that microorganisms grew better in com-plex media using yeast extract and peptone as nitrogen sources than in mineral media (Hahn-Hägerdal et al.,2005;La Grange et al.,1996).Some researchers also confirmed that yeast extract and peptone could increase the secretion and accumulation of het-erologous protein and inhibit the degradation of heterologous pro-tein (Macauley-Patrick et al.,2005;Wu et al.,2008).But yeast extract and peptone were expensive and the cost in fermentation medium greatly increased.Corn steep liquor powder,as a cheap raw material for industrial fermentation,containing plentiful nutrients,was the best alternative to extract and peptone for cell growth and heterologous protein expression of P.pastoris .The re-sults in shaken flasks and a 5-L fermenter also showed that CSD medium was the optimal medium for P.pastoris with low cost and simple contents.3.5.Analysis of CSD medium applied in industrial fermentation The total cost of large-scale industrial fermentation includes cap-ital cost,carbon source,nitrogen source,other medium source,power,steam,aseptic air,labor,maintenance,etc.The feedstocks ac-counted for about 30%of the total production cost (Hahn-Hägerdal et al.,2005;Lee,2005).In Chinese market,the price of corn steep li-quor powder was about 314–470USD/ton,and the price of indus-trial yeast extract and peptone was about 1567USD/ton.Prices of industrial inorganic salts used in BSM medium also were higher than that of corn steep liquor powder.Based on the available price infor-mation in Chinese market (Table 3),the costs of main components except glucose were about 6.4,20.0and 32.3USD/ton of culture with CSD,BSM and YPD media,respectively.This indicated that the cost with CSD medium was far lower than with YPD andBSMfed-batch fermentation in a 5-L fermenter using CSD medium and a novel fed-batch strategy combining the real-time L initial medium and 2.0L feeding volume.Dry wet weight (Ç),activity (j ),the concentration of glucose (N ),dissolved determined.Table 2Comparison of parameters for b -mananase production in a 5-L fermenter using YPD,BSM and CSD media based on the novel fed-batch strategy.Culture medium Time (h)a Overall specific growth rate,l (h À1)b Maximum DCW (g/L)Activity (U/mL)Activityproductivity (U/mL h)c CSD 360.07853.52195.561.0YPD 610.04658.02188.835.9BSM810.02849.92029.224.4a Refers to the fermentation time for obtaining the highestb -mananase activity.bOverall specific growth rate was calculated based on the formula:l =ln2/t d ,in which t d referred to the biomass doubling time.cActivity productivity (U/mL h)=The highest b -mananase activity (U/mL)/the fermentation time for obtaining the highest b -mananase activity (h).。

芦佳琪,吴玉珍,张瑞,等. 基于HS-SPME-GC-MS 与电子鼻分析芹菜贮藏期间挥发性物质的变化[J]. 食品工业科技,2024,45(5):212−222. doi: 10.13386/j.issn1002-0306.2023040101LU Jiaqi, WU Yuzhen, ZHANG Rui, et al. Change of the Volatile Compounds from Celery Leaves during Storage Based on HS-SPME-GC-MS and E-nose[J]. Science and Technology of Food Industry, 2024, 45(5): 212−222. (in Chinese with English abstract). doi:10.13386/j.issn1002-0306.2023040101· 分析检测 ·基于HS-SPME-GC-MS 与电子鼻分析芹菜贮藏期间挥发性物质的变化芦佳琪1,吴玉珍1,张 瑞1,韩晶晶1,熊爱生2,郁志芳1, *(1.南京农业大学食品科技学院,江苏南京 210095;2.南京农业大学园艺学院,江苏南京 210095)摘 要:采用顶空固相微萃取技术结合气相色谱-质谱联用(headspace solid phase microextraction-gas chromato-graphy-mass spectrometry ,HS-SPME-GC-MS )和电子鼻技术分析了20.0 ℃贮藏期间芹菜叶片挥发性物质的组成和含量的变化。
结果显示,采用HS-SPME-GC-MS 技术从芹菜中共检测到108种挥发性物质,单萜类(43.2%~52.92%)和苯酞类(19.93%~28.97%)为主要组分,其中D-柠檬烯含量丰富(6600.64~48566.12 μg/kg )。

(英汉对照)分子生物学--名词解释α helix α螺旋A helical secondary structure in proteins.Pl. α helices. 蛋白质中一种螺旋形的二级结构。
复数:α helices。
α-amanitin α鹅膏蕈碱A toxin that inhibits the three eukaryotic RNA polymerases to different extents. Name derives from mushroom of genus Amanita in which toxin is found. 一种能不同程度地抑制三种真核生物RNA聚合酶的毒素。
名称来自于产生此毒素的Amanita属蘑菇。
β-galactosidase β-半乳糖苷酶Enzyme that cleaves lactose into galactose and glucose. Name origin: the bond cut by this enzyme is called a β-galactosidic bond. 将乳糖分解为半乳糖和葡萄糖的酶。
名称来源:该酶切割的键称为β-半乳糖苷键。
β sheet β折叠A secondary structure in proteins, relatively flat and formed hydrogen bonding between two parallel or anti-parallel stretches of polypeptide. 蛋白质的一种二级结构,相对平坦,在两条平行的或反向平行的肽段之间形成氢键。
σ subunit σ亚基Component of prokaryotic RNA polymerase holoenzyme. Required for recognition of promoters. 原核生物RNA聚合酶全酶的组成成分。

分子生物学重要概念AAbundance (mRNA 丰度):指每个细胞中mRNA 分子的数目。
Abundant mRNA(高丰度mRNA):由少量不同种类mRNA组成,每一种在细胞中出现大量拷贝。
Acceptor splicing site (受体剪切位点):内含子右末端和相邻外显子左末端的边界。
Acentric fragment(无着丝粒片段):(由打断产生的)染色体无着丝粒片段缺少中心粒,从而在细胞分化中被丢失。
Active site(活性位点):蛋白质上一个底物结合的有限区域。
Allele(等位基因):在染色体上占据给定位点基因的不同形式。
Allelic exclusion(等位基因排斥):形容在特殊淋巴细胞中只有一个等位基因来表达编码的免疫球蛋白质。
Allosteric control(别构调控):指蛋白质一个位点上的反应能够影响另一个位点活性的能力。
Alu-equivalent family(Alu 相当序列基因):哺乳动物基因组上一组序列,它们与人类Alu家族相关。
Alu family (Alu家族):人类基因组中一系列分散的相关序列,每个约300bp长。
每个成员其两端有Alu 切割位点(名字的由来)。
α-Amanitin(鹅膏覃碱):是来自毒蘑菇Amanita phalloides 二环八肽,能抑制真核RNA聚合酶,特别是聚合酶II 转录。
Amber codon (琥珀密码子):核苷酸三联体UAG,引起蛋白质合成终止的三个密码子之一。
Amber mutation (琥珀突变):指代表蛋白质中氨基酸密码子占据的位点上突变成琥珀密码子的任何DNA 改变。
Amber suppressors (琥珀抑制子):编码tRNA的基因突变使其反密码子被改变,从而能识别UAG 密码子和之前的密码子。
Aminoacyl-tRNA (氨酰-tRNA):是携带氨基酸的转运RNA,共价连接位在氨基酸的NH2基团和tRNA 终止碱基的3¢或者2¢-OH 基团上。

中英文对照的分子育种相关名词3'untranslated region (3'UTR) 3'非翻译区5'untranslated region (5; UTR) 5'非翻译区A chromosome A 染色体AATAAA 多腺苷酸化信号aberration 崎变abiogenesis 非生源说accessory chromosome 副染色体accessory nucleus 副核accessory protein 辅助蛋白accident variance 偶然变异Ac-Ds system Ac-Ds 系统acentric chromosome 无着丝粒染色体acentric fragment 无着丝粒片段acentric ring 无着丝粒环achromatin 非染色质acquired character 获得性状acrocentric chromosome 近端着丝粒染色体acrosyndesis 端部联会activating transcription factor 转录激活因子activator 激活剂activator element 激活单元activator protein( AP)激活蛋白activator-dissociation system Ac-Ds 激活解离系统active chromatin 活性染色质active site 活性部位adaptation 适应adaptive peak 适应高峰adaptive surface 适应面addition 附加物addition haploid 附加单倍体addition line 附加系additive effect 加性效应additive gene 加性基因additive genetic variance 加性遗传方差additive recombination 插人重组additive resistance 累加抗性adenosine 腺昔adenosine diphosphate (ADP )腺昔二鱗酸adenosine triphosphate( ATP)腺昔三憐酸adjacent segregation 相邻分离A-form DNA A 型DNAakinetic chromosome 无着丝粒染色体akinetic fragment 无着丝粒片断alien addition monosomic 外源单体生物alien chromosome substitution 外源染色体代换alien species 外源种alien-addition cell hybrid 异源附加细胞杂种alkylating agent 焼化剂allele 等位基因allele center 等位基因中心allele linkage analysis 等位基因连锁分析allele specific oligonucleotide(ASO)等位基因特异的寡核苷酸allelic complement 等位(基因)互补allelic diversity 等位(基因)多样化allelic exclusion 等位基因排斥allelic inactivation 等位(基因)失活allelic interaction 等位(基因)相互作用allelic recombination 等位(基因)重组allelic replacement 等位(基因)置换allelic series 等位(基因)系列allelic variation 等位(基因)变异allelism 等位性allelotype 等位(基因)型allohaploid 异源单倍体allopatric speciation 异域种alloploidy 异源倍性allopolyhaploid 异源多倍单倍体allopolyploid 异源多倍体allosyndesis 异源联会allotetraploid 异源四倍体alloheteroploid 异源异倍体alternation of generation 世代交替alternative transcription 可变转录alternative transcription initiation 可变转录起始Alu repetitive sequence, Alu family Alu 重复序列,Alu 家族ambiguous codon 多义密码子ambisense genome 双义基因组ambisense RNA 双义RNAaminoacyl-tRNA binding site氨酰基tRNA接合位点aminoacyl-tRNA synthetase 氨酰基tRNA连接酶amixis 无融合amorph 无效等位基因amphipolyploid 双多倍体amplicon 扩增子amplification 扩增amplification primer 扩增引物analysis of variance 方差分析anaphase (分裂)后期anaphase bridge (分裂)后期桥anchor cell 锚状细胞androgamete 雄配子aneuhaploid 非整倍单倍体aneuploid 非整倍体animal genetics 动物遗传学annealing 复性antibody 抗体anticoding strand 反编码链anticodon 反密码子anticodon arm 反密码子臂anticodon loop 反密码子环antiparallel 反向平行antirepressor 抗阻抑物antisense RNA 反义RNAantisense strand 反义链apogamogony 无融合结实apogamy 无配子生殖apomixis 无融合生殖arm ratio (染色体)臂比artificial gene人工基因artificial selection 人工选择asexual hybridization 无性杂交asexual propagation 无性繁殖asexual reproduction 无性生殖assortative mating 选型交配asynapsis 不联会asynaptic gene 不联会基因atavism 返祖atelocentric chromosome 非端着丝粒染色体attached X chromosome 并连X 染色体attachment site 附着位点attenuation 衰减attenuator 衰减子autarchic gene 自效基因auto-alloploid 同源异源体autoallopolyploid 同源异源多倍体autobivalent 同源二阶染色体auto-diploid 同源二倍体;自体融合二倍体autodiploidization 同源二倍化autoduplication 自体复制autogenesis自然发生autogenomatic 同源染色体组autoheteroploidy 同源异倍性autonomous transposable element 自主转座单元autonomously replicating sequence(ARS)自主复制序列autoparthenogenesis 自发单性生殖autopolyhaploid 同源多倍单倍体autopolyploid 同源多倍体autoradiogram 放射自显影图autosyndetic pairing 同源配对autotetraploid 同源四倍体autozygote 同合子auxotroph 营养缺陷体B chromosome B 染色体B1,first backcross generation 回交第一代B2,second backcross generation 回交第二代back mutation 回复突变backcross 回交backcross hybrid 回交杂种backcross parent 回交亲本backcross ratio 回交比率background genotype 背景基因型bacterial artification chromosome( BAC )细菌人工染色体Bacterial genetics 细菌遗传学Bacteriophage 噬菌体balanced lethal 平衡致死balanced lethal gene 平衡致死基因balanced linkage 平衡连锁balanced load 平衡负荷balanced polymorphism 平衡多态现象balanced rearrangements 平衡重组balanced tertiary trisomic 平衡三级三体balanced translocation 平衡异位balancing selection 平衡选择band analysis 谱带分析banding pattern (染色体)带型basal transcription apparatus 基础转录装置base analog 碱基类似物base analogue 类減基base content 减基含量base exchange 碱基交换base pairing mistake 碱基配对错误base pairing rules 碱基配对法则base substitution 减基置换base transition 减基转换base transversion 减基颠换base-pair region 碱基配对区base-pair substitution 碱基配对替换basic number of chromosome 染色体基数behavioral genetics 行为遗传学behavioral isolation 行为隔离bidirectional replication 双向复制bimodal distribution 双峰分布binary fission 二分裂binding protein 结合蛋白binding site 结合部位binucleate phase 双核期biochemical genetics 生化遗传学biochemical mutant 生化突变体biochemical polymorphism 生化多态性bioethics 生物伦理学biogenesis 生源说bioinformatics 生物信息学biological diversity 生物多样性biometrical genetics 生物统计遗传学(简称生统遗传学) bisexual reproduction 两性生殖bisexuality 两性现象bivalent 二价体blending inheritance 混合遗传blot transfer apparatus 印迹转移装置blotting membrane 印迹膜bottle neck effect 瓶颈效应branch migration 分支迁移breed variety 品种breeding 育种,培育;繁殖,生育breeding by crossing 杂交育种法breeding by separation 分隔育种法breeding coefficient 繁殖率breeding habit 繁殖习性breeding migration 生殖回游,繁殖回游breeding period 生殖期breeding place 繁殖地breeding population 繁殖种群breeding potential繁殖能力,育种潜能breeding range 繁殖幅度breeding season 繁殖季节breeding size 繁殖个体数breeding system 繁殖系统breeding true 纯育breeding value 育种值broad heritability 广义遗传率bulk selection 集团选择C0,acentric 无着丝粒的Cl,monocentric 单着丝粒C2, dicentric双着丝粒的C3,tricentric 三着丝粒的candidate gene 候选基因candidate-gene approach 候选基因法Canpbenmodel坎贝尔模型carytype染色体组型,核型catabolite activator protein 分解活化蛋白catabolite repression 分解代谢产物阻遏catastrophism 灾变说cell clone 细胞克隆cell cycle 细胞周期cell determination 细胞决定cell division 细胞分裂cell division cycle gene(CDC gene) 细胞分裂周期基因ceU division lag细胞分裂延迟cell fate 细胞命运cell fusion 细胞融合cell genetics 细胞的遗传学cell hybridization 细胞杂交cell sorter细胞分类器cell strain 细胞株cell-cell communication 细胞间通信center of variation 变异中心centimorgan(cM) 厘摩central dogma 中心法则central tendency 集中趋势centromere DNA 着丝粒DNAcentromere interference 着丝粒干扰centromere 着丝粒centromeric exchange ( CME)着丝粒交换centromeric inactivation 着丝粒失活centromeric sequence( CEN sequence)中心粒序列character divergence 性状趋异chemical genetics 化学遗传学chemigenomics 化学基因组学chiasma centralization 交叉中化chiasma terminalization 交叉端化chimera异源嵌合体Chi-square (x2) test 卡方检验chondriogene 线粒体基因chorionic villus sampling 绒毛膜取样chromatid abemition染色单体畸变chromatid break染色单体断裂chromatid bridge 染色单体桥chromatid interchange 染色单体互换chromatid interference 染色单体干涉chromatid tetrad 四分染色单体chromatid translocation 染色单体异位chromatin agglutination 染色质凝聚chromosomal aberration 染色体崎变chromosomal assignment 染色体定位chromosomal banding 染色体显带chromosomal disorder 染色体病chromosomal elimination 染色体消减chromosomal inheritance 染色体遗传chromosomal interference 染色体干扰chromosomal location 染色体定位chromosomal locus 染色体位点chromosomal mutation 染色体突变chromosomal pattern 染色体型chromosomal polymorphism 染色体多态性chromosomal rearrangement 染色体质量排chromosomal reproduction 染色体增殖chromosomal RNA 染色体RNA chromosomal shift 染色体变迁,染色体移位chromosome aberration 染色体畸变chromosome arm 染色体臂chromosome banding pattern 染色体带型chromosome behavior 染色体动态chromosome blotting 染色体印迹chromosome breakage 染色体断裂chromosome bridge 染色体桥chromosome coiling 染色体螺旋chromosome condensation 染色体浓缩chromosome constriction 染色体缢痕chromosome cycle 染色体周期chromosome damage 染色体损伤chromosome deletion 染色体缺失chromosome disjunction 染色体分离chromosome doubling 染色体加倍chromosome duplication 染色体复制chromosome elimination染色体丢失chromosome engineering 染色体工程chromosome evolution 染色体进化chromosome exchange 染色体交换chromosome fusion 染色体融合chromosome gap 染色体间隙chromosome hopping 染色体跳移chromosome interchange 染色体交换chromosome interference 染色体干涉chromosome jumping 染色体跳查chromosome knob 染色体结chromosome loop 染色体环chromosome lose染色体丢失chromosome map 染色体图chromosome mapping 染色体作图chromosome matrix 染色体基质chromosome mutation染色体突变chromosome non-disjunction染色体不分离chromosome paring染色体配对chromosome polymorphism 染色体多态性chromosome puff染色体疏松chromosome rearrangement染色体质量排chromosome reduplication 染色体再加倍chromosome repeat染色体质量叠chromosome scaffold 染色体支架chromosome segregation 染色体分离chromosome set 染色体组chromosome stickiness染色体粘性chromosome theory of heredity 染色体遗传学说chromosome theory of inheritance 染色体遗传学说chromosome thread 染色体丝chromosome walking 染色体步查chromosome-mediated gene transfer 染色体中介基因转移chromosomology 染色体学CIB method CIB法;性连锁致死突变出现频率检测法circular DNA 环林DNAcis conformation 顺式构象cis dominance 顺式显性cis-heterogenote顺式杂基因子cis-regulatory element 顺式调节兀件cis-trans test 顺反测验cladogram 进化树cloning vector 克隆载体C-meiosis C减数分裂C-metaphase C 中期C-mitosis C有丝分裂code degeneracy 密码简并coding capacity 编码容量coding ratio 密码比coding recognition site 密码识别位置coding region 编码区coding sequence 编码序列coding site 编码位置coding strand 密码链coding triplet 编码三联体codominance 共显性codon bias 密码子偏倚codon type 密码子型coefficient of consanguinity 近亲系数coefficient of genetic determination 遗传决定系数coefficient of hybridity 杂种系数coefficient of inbreeding 近交系数coefficient of migration 迁移系数coefficient of relationship 亲缘系数coefficient of variability 变异系数coevolution 协同进化coinducer 协诱导物cold sensitive mutant 冷敏感突变体colineartiy 共线性combining ability 配合力comparative genomics 比较基因组学competence 感受态competent cell感受态细胞competing groups 竞争类群competition advantage 竞争优势competitive exclusion principle 竞争排斥原理complementary DNA (cDNA)互补DNA complementary gene 互补基因complementation test 互补测验complete linkage 完全连锁complete selection 完全选择complotype 补体单元型composite transposon 复合转座子conditional gene 条件基因conditional lethal 条件致死conditional mutation 条件突变consanguinity 近亲consensus sequence 共有序列conservative transposition 保守转座constitutive heterochromatin 组成型染色质continuous variation 连续变异convergent evolution 趋同进化cooperativity 协同性coordinately controlled genes 协同控制基因core promoter element 核心启动子core sequence 核心序列co-repressor协阻抑物correlation coefficient相关系数cosegregation 共分离cosuppression 共抑制cotranfection 共转染cotranscript共转录物cotranscriptional processing共转录过程cotransduction 共转导cotransformation 共转化cotranslational secrection 共翻译分泌counterselection 反选择coupling phase 互引相covalently closed circular DNA(cccDNA)共价闭合环状DNA covariation 相关变异criss-cross inheritance 交叉遗传cross 杂交crossability 杂交性crossbred 杂种cross-campatibility 杂交亲和性cioss-infertility 杂交不育性crossing over 交换crossing-over map 交换图crossing-over value 交换值crossover products 交换产物crossover rates 交换率crossover reducer 交换抑制因子crossover suppressor 交换抑制因子crossover unit 交换单位crossover value 值crossover-type gamete 交换型配子C-value paradox C 值悖论cybrid 胞质杂种cyclin 细胞周期蛋白cytidme 胞苷cytochimera 细胞嵌合体cytogenetics 细胞遗传学cytohet 胞质杂合子cytologic 细胞学的cytological map 细胞学图cytoplasm细胞质cytoplasmic genome 胞质基因组cytoplasmic heredity 细胞质遗传cytqplasmic incompatibility 细胞质不亲和性cytoplasmic inheritance 细胞质遗传cytoplasmic male sterility 细胞质雄性不育cytoplasmic mutation 细胞质突变cytofdasmic segregation 细胞质分离cytoskeleton 细胞骨架Darwin 达尔文Darwinian fitness 达尔文适合度Darwinism 达尔文学说daughter cell 子细胞daughter chromatid 子染色体daughter chromosome 子染色体deformylase 去甲酰酶degenerate code 简并密码degenerate primer 简并引物degenerate sequence 简并序列degenerated codon 简并密码子degeneration 退化degree of dominance 显性度delayed inheritance 延迟遗传deletant 缺失体deletion 缺失deletion loop 缺失环deletion mapping 缺失作图deletion mutation 缺失突变denatured DNA 变性DNA denatured protein 变性蛋白denaturing gel 变性胶denaturing gel electrophoresis 变性凝胶电泳denaturing gradient polyacrylamide gel 变性聚丙稀酰胺凝胶density gradient centrifugation 密度梯度离心density gradient separation 密度梯度分离deoxyribonucleic acid-dependent DNA polymerase 依赖于DNA的DNA聚合酶derived line 衍生系derived type 衍生类型developmental genetics 发育遗传学developmental pathway 发育途径dicentric bridge 双粒染色体桥dicentric chromosome 双着丝粒染色体differential staining technique 显带技术differentiation center 分化中心dihaploid 双单倍体,dihybrid 双因子杂种dihybrid cross 双因子杂交dimorphism 二态性diploidization 二倍化diploidize 二倍化diploidized haploid 二倍化的单倍体direct cross 正交direct repeat 同向重复(序列)direct selection 正选择directed mutagenesis 正向突变discontinuous variation 不连续变异distant hybrid 远缘杂种distant hybridization 远缘杂交diversity center 多样性中心diversity curve 多样性曲线diversity gene ( D gene) D 基因diversity indices 多样性指数diversity of species 种的多样性diversity region ( D region) D 区;多变区DNA alkylation DNA 烧化DNA amplification DNA 扩增DNA amplification in vitro DNA 体外扩增DNA amplification polymorphism DNA 扩增多态性DNA breakage DNA 断裂DNA database DNA 数据库DNA degradation DNA 降解DNA denaturation DNA 变性DNA detection DNA 检测DNA distortion DNA 变形DNA duplex DNA 双链体DNA duplicase DNA 复合酶DNA element DNA 单元DNA evolution DNA 进化DNA fingerprint DNA 指纹DNA fingerprinting DNA 指纹分析DNA homology DNA 同源性DNA hybridization DNA 杂交DNA jumping technique DNA 跳查技术DNA melting DNA 解链DNA methylation DNA 甲基化DNA modification DNA 修饰DNA modification restriction system DNA 修饰限制系统DNA nicking DNA 切口形成DNA oxidation DNA 氧化DNA packaging DNA 包装DNA pairing DNA 配对DNA pitch DNA 螺距DNA polymorphism DNA 多态性DNA probe DNA 探针DNA puff DNA 泡DNA purification DNA 纯化DNA recombination DNA 重组DNA redundant 多余DNADNA repair DNA 修复DNA replication DNA 复制DNA replication enhancer DNA 复制增强子DNA replication origin DNA 复制起点DNA replication site DNA 复制点DNA sealase DNA 连接酶DNA sequence analysis DNA 序列分析DNA sizing gene DNA大小决定基因DNA strand exchange DNA 链交换DNA strand separation DNA 链分离DNA strand transfer protein DNA 链转移蛋白DNA template DNA 模板DNA thermal cycler DNA 热循环仪DNA topoisomerase DNA 拓扑异构酶DNA transcript DNA 转录物DNA transposon DNA 转座子DNA twist DNA 扭曲DNA typing DNA 分型DNA untwisting DNA 解旋DNA unwinding enzyme DNA 解旋酶DNA unwinding protein DNA 解旋蛋白DNA-agar technique DNA 琼脂技术DNAase I footprinting DNA 酶I 足迹法DNAase-free reagent 无DNA 酶试剂DNA-binding domain DNA 结合域DNA-binding motif DNA 结合基序DNA-binding protein DNA 结合蛋白DNA-polymerase DNA 聚合酶DNA-protein complex DNA -蛋白质复合体DNA-protein interaction DNA _ 蛋白质相互作用DNA-restriction enzyme DNA 限制酶DNA-RNA hybrid DNA-RNA 杂交体DNase-free 不含DNA 酶的dominance 显性dominance type 优势型dominance variance 显性方差dominant allele 显性等位基因dominant effect 显性效应dominant gene 显性基因dominant gene mutation 显'性基因突变dominant lethal 显性致死dominant phenotype 显性表型donor DNA 供体DNAdonor organism 供体生物dosage compensation 剂量补偿作用dotting blotting 点溃法double crossing over 双交换double fertilization 汉受精duplicate genes 重复基因duplication重复duplicon 重复子dyad 二分体dynamic selection 动态选择ecological genetics 生态遗传学ecological isolation 生态隔离ecological niche 生态小境ectopic expression 异位表达ectopic integration 异位整合effective population size 有效群体大小embryoid 胚状体embryonic stem cells( ES cells)胚胎干细胞endocrine signal 内分泌信号endogamy 近亲繁殖endomitosis 核内有丝分裂endonuclease 内切核酸酶endopolyploidy 核内多倍体environment 环境environmental variance 环境方差environmental variation 环境变异epigenesis 后成说epigenetic inheritance 后生遗传epigenetically silenced 后生沉默episome 附加体epistasis 上位性epistatic dominance 超显性epistatic gene 上位基因equal segregation 均等分离equational division 均等分裂equilibrium population 平衡群体Expressed Sequence Tag(EST)表达序列标签euchromatin 常染色质euchromatin常染色质eugenics 优生学euhaploid 整单倍体eukaryote 真核生物eukaryotic chromosome 真核染色体eukaryotic cell 真核细胞eukaryotic organism 真核生物eukaryotic vector 真核载体euphenics 优型学euploid 整倍体evolutional load 进化负荷evolutionary divergence 进化趋异evolutionary genetics 进化遗传学evolutionaiy rate 进化速率excision repair 切除修复exconjugant 接合后体excretion vector 分泌型载体exit site 萌发点exogenote 外基因子exogenous gene 外源基因exonuclease 外切核酸酶expression cloning 表达克隆expression library 表达文库expression mutation 表达突变expression plasmid 表达质粒expression product 表达产物expression screening 表达筛选extinguisher loci 消失基因座,灭绝基因座extirpated species 绝迹种extrachromosomal inheritance 染色体外遗传extra-chromosome超数染色体,额外染色体extranuclear inheritance 核外遗传F1 generation F1代,子一代F2 generation F2 代,子二代facultative heterochromatin 兼性异染色质familial trait 家族性状family selection 家系选择feedback suppression 反馈抑制female gamete 雌配子fertility factor 致育因子filial generation 子代fingerprint 指纹finite population 有限群体first division segregation 第一次分裂分离first division segregation pattern 第一次分裂分离模式flanking sequence 侧翼序列flow cytometry 流式细胞仪fluorescence in situ hybridization ( FISH )荧光原位杂交fluorescent primer 荧光引物fluorescent probe 荧光探针formyl methionine (fMet)甲酰甲硫氨酸foot printing 足迹法foreign DNA 外源DNAforward genetics 正向遗传学forward mutation 正向突变forward primer 正向引物founder effect 建立者效应four strand double crossing over 四线双交换full-sib 全同胞functional genomics 功能基因组学functional RNA 功能RNAgain-of-function mutation 功能获得性突变gamete 配子gametic 配子的gametic incompatibility 配子不亲和性gametic lethal 配子致死gametic linkage 配子连锁gametic meiosis 配子减数分裂gametic ratio 配子分离比gametoclonal variation 配子无性系变异gametophyte 配子体G-band G带;中期染色体带GC box GC 框GC tailing GC 加尾gel electrophoresis 凝胶电泳gemetic sterility 配子不育gene activation 基因激活gene activity 基因活性gene amplification 基因扩增gene analysis 基因分析gene arrangement 基因排列gene balance 基因平衡gene basis 基因基础gene batteries 基因群gene block 基因区段gene carrier 基因携带者gene center theory 基因中心学说gene cluster 基因簇gene combination 基因重组gene complex 基因复合体gene content 基因含量gene conversion 基因转换gene distribution 基因分布gene diversity 基因多样性gene dosage 基因剂量gene dosage compensation 基因剂量补偿gene dosage effect 基因剂量效应gene duplication 基因重复gene element 基因元件gene exchange 基因交流gene expression 基因表达gene expression system 基因表达系统gene family 基因家族gene fixation 基因固定gene flow 基因流gene frequency 基因频率gene fusion 基因融合gene inactivation 基因失活gene inoculation 基因接种gene interaction 基因相互作用gene isolation 基因分离gene knockout 基因敲除gene knock-out 基因失效法gene linkage 基因连锁gene localization 基因定位gene location 基因位置gene locus 基因位点gene magnification 基因扩增gene manipulation 基因操作gene map 基因图谱gene mapping 基因作图gene multiplication 基因重复gene mutation 基因突变gene mutation rate 基因突变频率gene order 基因次序gene organization 基因组构gene pool 基因库gene position effect 基因位置效应gene probe 基因探针gene product 基因产物gene rearrangement 某因重排gene reassortment 基因重新配对gene replication 基因复制gene repression 基因抑制gene resortment 基因重配gene silencing 基因沉默gene splicing 基因剪接gene string 基因线gene structure 基因结构gene substitute 基因置换gene substitution 基因置换gene suppression 基因抑制gene synthesis 基因合成gene tagged 基因标签gene tagging 基因标签gene targeting 基因导向,基因寻靶gene transfer 基因转移gene transfer agent 基因传递因子gene transfer vector 基因转移载体gene transposition 基因转座genealogical classification 系谱分类genera 属general transcription factor ( GTF )通用转录因子generalized transduction 普遍性转导generation 世代generative cell 生殖细胞generative reproduction 有性繁殖generic coefficient 种属系数generic cross 属间杂交generic name 属名genes in common 共同基因gene-specific transcription factor 基因特异性转录因子genetic ablation 基因缺损genetic advance 遗传进度genetic algebra 遗传代数genetic analysis 遗传分析genetic background 遗传背景genetic balance 遗传平衡genetic block 遗传性阻碍genetic compensation 遗传补偿genetic complementation 遗传互补genetic composition 遗传组成genetic continuity 遗传连续性genetic control 遗传控制genetic covariance 遗传协方差genetic cross 杂交genetic database 遗传数据库genetic death 遗传性死亡genetic deficiency 遗传缺损genetic deformity 基因变型genetic determinant 遗传决定因子genetic dimorphism 遗传二型现象genetic distance 遗传距离genetic divergence 遗传趋异genetic diversity 遗传多样性genetic dominance 遗传优势genetic donor 基因供体genetic drift 遗传漂变genetic element遗传因子,遗传成分genetic engineering 遗传工程genetic equilibrium 遗传平衡genetic erosion 遗传冲刷,遗传蚀变genetic expression 遗传表达genetic extinction 遗传灭绝genetic facilitation 遗传促进作用genetic factor 遗传因子genetic feedback 遗传反馈genetic fingerprint 遗传指纹genetic fingerprinting 遗传指纹分析genetic fitness 遗传适合度genetic flexibility 遗传可塑性genetic gain 遗传获得量genetic heterogeneity 遗传异质性genetic homology 遗传同源genetic immunity 遗传免疫genetic imprinting 遗传印记genetic inertia 遗传惰性genetic information 遗传信息genetic inoculation 基因接种genetic instability 遗传不稳定性genetic continuity 遗传连续性genetic control 遗传控制genetic covariance 遗传协方差genetic cross 杂交genetic database 遗传数据库genetic death 遗传性死亡genetic deficiency 遗传缺损genetic deformity 基因变型genetic determinant 遗传决定因子genetic dimorphism 遗传二型现象genetic distance 遗传距离genetic divergence 遗传趋异genetic diversity 遗传多样性genetic dominance 遗传优势genetic donor 基因供体genetic drift 遗传漂变genetic element遗传因子,遗传成分genetic engineering 遗传工程genetic equilibrium 遗传平衡genetic erosion 遗传冲刷,遗传蚀变genetic expression 遗传表达genetic extinction 遗传灭绝genetic facilitation 遗传促进作用genetic factor 遗传因子genetic feedback 遗传反馈genetic fingerprint 遗传指纹genetic fingerprinting 遗传指纹分析genetic fitness 遗传适合度genetic flexibility 遗传可塑性genetic gain 遗传获得量genetic heterogeneity 遗传异质性genetic homology 遗传同源genetic immunity 遗传免疫genetic imprinting 遗传印记genetic inertia 遗传惰性genetic information 遗传信息genetic inoculation 基因接种genetic instability 遗传不稳定性genetic interaction 遗传相互作用genetic isolating factor 遗传隔离因子genetic isolation 遗传隔离genetic knock-out experiment 基因失效试验genetic linkage 遗传连锁genetic linkage map 遗传连锁图谱genetic load 遗传负荷genetic manipulation 遗传操作genetic map 遗传图谱genetic mapping 遗传作图genetic marker 遗传标记genetic masking 基因组掩饰genetic material 遗传物质genetic mobilization 遗传转移genetic modification 遗传修饰genetic module 遗传组件genetic nomenclature 遗传命名法genetic parameter 遗传参数genetic polarity 遗传极性genetic polymorphism 遗传多样性genetic population 遗传群体genetic potential 遗传潜力genetic process 遗传过程genetic property 遗传特'性genetic ratio 遗传比genetic reactivation 遗传复活genetic reassortment 遗传重排genetic recipient 基因受体genetic recombination 遗传重组genetic regulation 遗传调节genetic relationship 亲缘关系genetic repair mechanism 遗传修复机制genetic replication 遗传复制genetic risk 遗传危险性genetic screening 遗传筛查genetic segregation 遗传分离genetic selection 遗传选择genetic sex 遗传性别genetic shift 遗传漂移genetic stability 遗传稳定性genetic sterility 遗传性不育genetic strain 遗传品系genetic suppression 遗传抑制genetic switch 遗传开关genetic system 遗传体系genetic transcription 遗传转录genetic transformation 遗传转换genetic translation 遗传翻译genetic transmission 遗传传递genetic typing 遗传分型genetic unit 遗传单位genetic value 遗传值genetic variability 遗传变异性genetic variance 遗传方差genetic vulnerability 遗传易损性genetic“hot spot” 遗传“热点”genetical marker 遗传标记genetical non-disjunction 遗传不分离genetical population 遗传群体genetically heterogeneous 遗传异质的genetically modified organism 基因修饰生物genetics correction 遗传修正genetics of resistance 抗性遗传genetype 基因型genic balance 基因平衡genome allopolyploid 基因组异质多倍体genome amplification 基因组扩增genome evolution 基因组进化genome mapping 基因组作图genome project 基因组计划genome rearrangement 基因组重排genome sequencing 基因组测序genomic exclusion 基因组排斥genomic fingerprinting 基因组指纹分析genomic footprinting 基因组足迹分析genomic imprinting 基因组印记genomic instability 基因组不稳定性genomic library 基因组文库genomic walking 基因组步查genotypic frequency 基因型频率genotypic ratio 基因型比值genotypic value 基因型值genotypic variance 基因型方差geographic speciation 地理型新种形成geographical isolation 地理隔离geographical polymorphism 地理多态现象germ layer 胚层germ line 种系germ nucleus 生殖核germ plasm 种质germinal mutation 生殖细胞突变germ-line gene therapy 种系基因治疗giant chromosome 巨型染色体global homology 总体同源性global region 全局调节子globular protein 球蛋白group selection 集团选择growth factor 生长因子GT-AG rule mRNA剪接识别信号规则gynandromorphy 雌雄嵌合体hairpin loop 发夹环hairpin structure 发夹结构half life 半寿期half sib mating 半同胞交配haplogenotypic 单倍基因型的haploid 单倍体haploidization 单倍体化haplotype 单元型hapostatic gene 下位基因Hardy-Weinberg equilibrium 哈迪-温伯格平衡heat shock gene 热激基因heat sock protein 热激蛋白heavy chain 重链helical structure 螺旋结构。

非等位基因概述非等位基因是指同一基因座上的不同等位基因。
等位基因是指在某个给定的基因座上,可以存在多种不同的变体。
每个个体继承了一对等位基因,一对等位基因可能会导致不同的表型表达。
非等位基因的存在使得遗传学研究更加复杂,因为不同的等位基因会对个体的表型产生不同的影响。
背景在生物学中,基因座是指染色体上一个特定的位置,该位置上的基因决定了某个特征的表达方式。
每个基因座上可以有多种不同的等位基因。
等位基因是指在某个特定基因座上的不同基因变体。
每个个体都会继承一对等位基因,通过这对等位基因的不同组合,决定了个体的表型。
然而,并非所有基因座上的等位基因都具有相同的表现型。
非等位基因的影响非等位基因的存在导致不同等位基因会对个体表型产生不同的影响。
有些非等位基因会表现出显性效应,也就是说,当个体继承了一个突变的等位基因时,即使同时继承了一个正常的等位基因,但显性效应会使得突变的等位基因的表型表达得到体现。
相反,有些非等位基因会表现出隐性效应,当个体继承了两个突变的等位基因时,才会表现出突变的表型。
除了显性和隐性效应之外,非等位基因还可能发生两种其他类型的表型效应。
一种是共显效应,当个体继承了两个不同的突变等位基因时,在表型表达上会表现出一种新的特征,这个特征并不是单个突变等位基因所能导致的。
另一种是部分显性效应,当个体继承了两个不同的突变等位基因时,表型表达将介于两个单独突变等位基因的表型之间。
重组和非等位基因重组是指两个不同的染色体交换部分基因序列的过程。
在重组的过程中,非等位基因可能会发生改变,导致新的等位基因组合形成。
这一过程使得非等位基因的表型效应更加复杂,因为新的等位基因可能将不同基因座的效应组合起来。
非等位基因的重要性非等位基因对生物的适应性和多样性起着重要作用。
通过对等位基因的各种组合的研究,人们可以更好地理解基因与表型之间的关系,并揭示遗传变异对物种适应环境的重要性。
总结非等位基因是指同一基因座上的不同等位基因。

Dynamic and distribution of ammonia-oxidizing bacteria communities during sludge granulation in an anaerobic e aerobic sequencing batch reactorZhang Bin a ,b ,Chen Zhe a ,b ,Qiu Zhigang a ,b ,Jin Min a ,b ,Chen Zhiqiang a ,b ,Chen Zhaoli a ,b ,Li Junwen a ,b ,Wang Xuan c ,*,Wang Jingfeng a ,b ,**aInstitute of Hygiene and Environmental Medicine,Academy of Military Medical Sciences,Tianjin 300050,PR China bTianjin Key Laboratory of Risk Assessment and Control for Environment and Food Safety,Tianjin 300050,PR China cTianjin Key Laboratory of Hollow Fiber Membrane Material and Membrane Process,Institute of Biological and Chemical Engineering,Tianjin Polytechnical University,Tianjin 300160,PR Chinaa r t i c l e i n f oArticle history:Received 30June 2011Received in revised form 10September 2011Accepted 10September 2011Available online xxx Keywords:Ammonia-oxidizing bacteria Granular sludgeCommunity development Granule sizeNitrifying bacteria distribution Phylogenetic diversitya b s t r a c tThe structure dynamic of ammonia-oxidizing bacteria (AOB)community and the distribution of AOB and nitrite-oxidizing bacteria (NOB)in granular sludge from an anaerobic e aerobic sequencing batch reactor (SBR)were investigated.A combination of process studies,molecular biotechniques and microscale techniques were employed to identify and characterize these organisms.The AOB community structure in granules was substantially different from that of the initial pattern of the inoculants sludge.Along with granules formation,the AOB diversity declined due to the selection pressure imposed by process conditions.Denaturing gradient gel electrophoresis (DGGE)and sequencing results demonstrated that most of Nitrosomonas in the inoculating sludge were remained because of their ability to rapidly adapt to the settling e washing out action.Furthermore,DGGE analysis revealed that larger granules benefit more AOB species surviving in the reactor.In the SBR were various size granules coexisted,granule diameter affected the distribution range of AOB and NOB.Small and medium granules (d <0.6mm)cannot restrict oxygen mass transfer in all spaces of the rger granules (d >0.9mm)can result in smaller aerobic volume fraction and inhibition of NOB growth.All these observations provide support to future studies on the mechanisms responsible for the AOB in granules systems.ª2011Elsevier Ltd.All rights reserved.1.IntroductionAt sufficiently high levels,ammonia in aquatic environments can be toxic to aquatic life and can contribute to eutrophica-tion.Accordingly,biodegradation and elimination of ammonia in wastewater are the primary functions of thewastewater treatment process.Nitrification,the conversion of ammonia to nitrate via nitrite,is an important way to remove ammonia nitrogen.It is a two-step process catalyzed by ammonia-oxidizing and nitrite-oxidizing bacteria (AOB and NOB).Aerobic ammonia-oxidation is often the first,rate-limiting step of nitrification;however,it is essential for the*Corresponding author .**Corresponding author.Institute of Hygiene and Environmental Medicine,Academy of Military Medical Sciences,Tianjin 300050,PR China.Tel.:+862284655498;fax:+862223328809.E-mail addresses:wangxuan0116@ (W.Xuan),jingfengwang@ (W.Jingfeng).Available online atjournal homepage:/locate/watresw a t e r r e s e a r c h x x x (2011)1e 100043-1354/$e see front matter ª2011Elsevier Ltd.All rights reserved.doi:10.1016/j.watres.2011.09.026removal of ammonia from the wastewater(Prosser and Nicol, 2008).Comparative analyses of16S rRNA sequences have revealed that most AOB in activated sludge are phylogeneti-cally closely related to the clade of b-Proteobacteria (Kowalchuk and Stephen,2001).However,a number of studies have suggested that there are physiological and ecological differences between different AOB genera and lineages,and that environmental factors such as process parameter,dis-solved oxygen,salinity,pH,and concentrations of free ammonia can impact certain species of AOB(Erguder et al., 2008;Kim et al.,2006;Koops and Pommerening-Ro¨ser,2001; Kowalchuk and Stephen,2001;Shi et al.,2010).Therefore, the physiological activity and abundance of AOB in waste-water processing is critical in the design and operation of waste treatment systems.For this reason,a better under-standing of the ecology and microbiology of AOB in waste-water treatment systems is necessary to enhance treatment performance.Recently,several developed techniques have served as valuable tools for the characterization of microbial diversity in biological wastewater treatment systems(Li et al., 2008;Yin and Xu,2009).Currently,the application of molec-ular biotechniques can provide clarification of the ammonia-oxidizing community in detail(Haseborg et al.,2010;Tawan et al.,2005;Vlaeminck et al.,2010).In recent years,the aerobic granular sludge process has become an attractive alternative to conventional processes for wastewater treatment mainly due to its cell immobilization strategy(de Bruin et al.,2004;Liu et al.,2009;Schwarzenbeck et al.,2005;Schwarzenbeck et al.,2004a,b;Xavier et al.,2007). Granules have a more tightly compact structure(Li et al.,2008; Liu and Tay,2008;Wang et al.,2004)and rapid settling velocity (Kong et al.,2009;Lemaire et al.,2008).Therefore,granular sludge systems have a higher mixed liquid suspended sludge (MLSS)concentration and longer solid retention times(SRT) than conventional activated sludge systems.Longer SRT can provide enough time for the growth of organisms that require a long generation time(e.g.,AOB).Some studies have indicated that nitrifying granules can be cultivated with ammonia-rich inorganic wastewater and the diameter of granules was small (Shi et al.,2010;Tsuneda et al.,2003).Other researchers reported that larger granules have been developed with the synthetic organic wastewater in sequencing batch reactors(SBRs)(Li et al., 2008;Liu and Tay,2008).The diverse populations of microor-ganisms that coexist in granules remove the chemical oxygen demand(COD),nitrogen and phosphate(de Kreuk et al.,2005). However,for larger granules with a particle diameter greater than0.6mm,an outer aerobic shell and an inner anaerobic zone coexist because of restricted oxygen diffusion to the granule core.These properties of granular sludge suggest that the inner environment of granules is unfavorable to AOB growth.Some research has shown that particle size and density induced the different distribution and dominance of AOB,NOB and anam-mox(Winkler et al.,2011b).Although a number of studies have been conducted to assess the ecology and microbiology of AOB in wastewater treatment systems,the information on the dynamics,distribution,and quantification of AOB communities during sludge granulation is still limited up to now.To address these concerns,the main objective of the present work was to investigate the population dynamics of AOB communities during the development of seedingflocs into granules,and the distribution of AOB and NOB in different size granules from an anaerobic e aerobic SBR.A combination of process studies,molecular biotechniques and microscale techniques were employed to identify and char-acterize these organisms.Based on these approaches,we demonstrate the differences in both AOB community evolu-tion and composition of theflocs and granules co-existing in the SBR and further elucidate the relationship between distribution of nitrifying bacteria and granule size.It is ex-pected that the work would be useful to better understand the mechanisms responsible for the AOB in granules and apply them for optimal control and management strategies of granulation systems.2.Material and methods2.1.Reactor set-up and operationThe granules were cultivated in a lab-scale SBR with an effective volume of4L.The effective diameter and height of the reactor was10cm and51cm,respectively.The hydraulic retention time was set at8h.Activated sludge from a full-scale sewage treat-ment plant(Jizhuangzi Sewage Treatment Works,Tianjin, China)was used as the seed sludge for the reactor at an initial sludge concentration of3876mg LÀ1in MLSS.The reactor was operated on6-h cycles,consisting of2-min influent feeding,90-min anaerobic phase(mixing),240-min aeration phase and5-min effluent discharge periods.The sludge settling time was reduced gradually from10to5min after80SBR cycles in20days, and only particles with a settling velocity higher than4.5m hÀ1 were retained in the reactor.The composition of the influent media were NaAc(450mg LÀ1),NH4Cl(100mg LÀ1),(NH4)2SO4 (10mg LÀ1),KH2PO4(20mg LÀ1),MgSO4$7H2O(50mg LÀ1),KCl (20mg LÀ1),CaCl2(20mg LÀ1),FeSO4$7H2O(1mg LÀ1),pH7.0e7.5, and0.1mL LÀ1trace element solution(Li et al.,2007).Analytical methods-The total organic carbon(TOC),NHþ4e N, NOÀ2e N,NOÀ3e N,total nitrogen(TN),total phosphate(TP) concentration,mixed liquid suspended solids(MLSS) concentration,and sludge volume index at10min(SVI10)were measured regularly according to the standard methods (APHA-AWWA-WEF,2005).Sludge size distribution was determined by the sieving method(Laguna et al.,1999).Screening was performed with four stainless steel sieves of5cm diameter having respective mesh openings of0.9,0.6,0.45,and0.2mm.A100mL volume of sludge from the reactor was sampled with a calibrated cylinder and then deposited on the0.9mm mesh sieve.The sample was subsequently washed with distilled water and particles less than0.9mm in diameter passed through this sieve to the sieves with smaller openings.The washing procedure was repeated several times to separate the gran-ules.The granules collected on the different screens were recovered by backwashing with distilled water.Each fraction was collected in a different beaker andfiltered on quantitative filter paper to determine the total suspended solid(TSS).Once the amount of total suspended solid(TSS)retained on each sieve was acquired,it was reasonable to determine for each class of size(<0.2,[0.2e0.45],[0.45e0.6],[0.6e0.9],>0.9mm) the percentage of the total weight that they represent.w a t e r r e s e a r c h x x x(2011)1e10 22.2.DNA extraction and nested PCR e DGGEThe sludge from approximately8mg of MLSS was transferred into a1.5-mL Eppendorf tube and then centrifuged at14,000g for10min.The supernatant was removed,and the pellet was added to1mL of sodium phosphate buffer solution and aseptically mixed with a sterilized pestle in order to detach granules.Genomic DNA was extracted from the pellets using E.Z.N.A.äSoil DNA kit(D5625-01,Omega Bio-tek Inc.,USA).To amplify ammonia-oxidizer specific16S rRNA for dena-turing gradient gel electrophoresis(DGGE),a nested PCR approach was performed as described previously(Zhang et al., 2010).30m l of nested PCR amplicons(with5m l6Âloading buffer)were loaded and separated by DGGE on polyacrylamide gels(8%,37.5:1acrylamide e bisacrylamide)with a linear gradient of35%e55%denaturant(100%denaturant¼7M urea plus40%formamide).The gel was run for6.5h at140V in 1ÂTAE buffer(40mM Tris-acetate,20mM sodium acetate, 1mM Na2EDTA,pH7.4)maintained at60 C(DCodeäUniversal Mutation Detection System,Bio-Rad,Hercules,CA, USA).After electrophoresis,silver-staining and development of the gels were performed as described by Sanguinetti et al. (1994).These were followed by air-drying and scanning with a gel imaging analysis system(Image Quant350,GE Inc.,USA). The gel images were analyzed with the software Quantity One,version4.31(Bio-rad).Dice index(Cs)of pair wise community similarity was calculated to evaluate the similarity of the AOB community among DGGE lanes(LaPara et al.,2002).This index ranges from0%(no common band)to100%(identical band patterns) with the assistance of Quantity One.The Shannon diversity index(H)was used to measure the microbial diversity that takes into account the richness and proportion of each species in a population.H was calculatedusing the following equation:H¼ÀPn iNlogn iN,where n i/Nis the proportion of community made up by species i(bright-ness of the band i/total brightness of all bands in the lane).Dendrograms relating band pattern similarities were automatically calculated without band weighting(consider-ation of band density)by the unweighted pair group method with arithmetic mean(UPGMA)algorithms in the Quantity One software.Prominent DGGE bands were excised and dissolved in30m L Milli-Q water overnight,at4 C.DNA was recovered from the gel by freeze e thawing thrice.Cloning and sequencing of the target DNA fragments were conducted following the estab-lished method(Zhang et al.,2010).2.3.Distribution of nitrifying bacteriaThree classes of size([0.2e0.45],[0.45e0.6],>0.9mm)were chosen on day180for FISH analysis in order to investigate the spatial distribution characteristics of AOB and NOB in granules.2mg sludge samples werefixed in4%para-formaldehyde solution for16e24h at4 C and then washed twice with sodium phosphate buffer;the samples were dehydrated in50%,80%and100%ethanol for10min each. Ethanol in the granules was then completely replaced by xylene by serial immersion in ethanol-xylene solutions of3:1, 1:1,and1:3by volume andfinally in100%xylene,for10min periods at room temperature.Subsequently,the granules were embedded in paraffin(m.p.56e58 C)by serial immer-sion in1:1xylene-paraffin for30min at60 C,followed by 100%paraffin.After solidification in paraffin,8-m m-thick sections were prepared and placed on gelatin-coated micro-scopic slides.Paraffin was removed by immersing the slide in xylene and ethanol for30min each,followed by air-drying of the slides.The three oligonucleotide probes were used for hybridiza-tion(Downing and Nerenberg,2008):FITC-labeled Nso190, which targets the majority of AOB;TRITC-labeled NIT3,which targets Nitrobacter sp.;TRITC-labeled NSR1156,which targets Nitrospira sp.All probe sequences,their hybridization condi-tions,and washing conditions are given in Table1.Oligonu-cleotides were synthesized andfluorescently labeled with fluorochomes by Takara,Inc.(Dalian,China).Hybridizations were performed at46 C for2h with a hybridization buffer(0.9M NaCl,formamide at the percentage shown in Table1,20mM Tris/HCl,pH8.0,0.01% SDS)containing each labeled probe(5ng m LÀ1).After hybrid-ization,unbound oligonucleotides were removed by a strin-gent washing step at48 C for15min in washing buffer containing the same components as the hybridization buffer except for the probes.For detection of all DNA,4,6-diamidino-2-phenylindole (DAPI)was diluted with methanol to afinal concentration of1ng m LÀ1.Cover the slides with DAPI e methanol and incubate for15min at37 C.The slides were subsequently washed once with methanol,rinsed briefly with ddH2O and immediately air-dried.Vectashield(Vector Laboratories)was used to prevent photo bleaching.The hybridization images were captured using a confocal laser scanning microscope (CLSM,Zeiss710).A total of10images were captured for each probe at each class of size.The representative images were selected andfinal image evaluation was done in Adobe PhotoShop.w a t e r r e s e a r c h x x x(2011)1e1033.Results3.1.SBR performance and granule characteristicsDuring the startup period,the reactor removed TOC and NH 4þ-N efficiently.98%of NH 4þ-N and 100%of TOC were removed from the influent by day 3and day 5respectively (Figs.S2,S3,Supporting information ).Removal of TN and TP were lower during this period (Figs.S3,S4,Supporting information ),though the removal of TP gradually improved to 100%removal by day 33(Fig.S4,Supporting information ).To determine the sludge volume index of granular sludge,a settling time of 10min was chosen instead of 30min,because granular sludge has a similar SVI after 60min and after 5min of settling (Schwarzenbeck et al.,2004b ).The SVI 10of the inoculating sludge was 108.2mL g À1.The changing patterns of MLSS and SVI 10in the continuous operation of the SBR are illustrated in Fig.1.The sludge settleability increased markedly during the set-up period.Fig.2reflects the slow andgradual process of sludge granulation,i.e.,from flocculentsludge to granules.3.2.DGGE analysis:AOB communities structure changes during sludge granulationThe results of nested PCR were shown in Fig.S1.The well-resolved DGGE bands were obtained at the representative points throughout the GSBR operation and the patterns revealed that the structure of the AOB communities was dynamic during sludge granulation and stabilization (Fig.3).The community structure at the end of experiment was different from that of the initial pattern of the seed sludge.The AOB communities on day 1showed 40%similarity only to that at the end of the GSBR operation (Table S1,Supporting information ),indicating the considerable difference of AOB communities structures between inoculated sludge and granular sludge.Biodiversity based on the DGGE patterns was analyzed by calculating the Shannon diversity index H as204060801001201401254159738494104115125135147160172188Time (d)S V I 10 (m L .g -1)10002000300040005000600070008000900010000M L S S (m g .L -1)Fig.1e Change in biomass content and SVI 10during whole operation.SVI,sludge volume index;MLSS,mixed liquid suspendedsolids.Fig.2e Variation in granule size distribution in the sludge during operation.d,particle diameter;TSS,total suspended solids.w a t e r r e s e a r c h x x x (2011)1e 104shown in Fig.S5.In the phase of sludge inoculation (before day 38),H decreased remarkably (from 0.94to 0.75)due to the absence of some species in the reactor.Though several dominant species (bands2,7,10,11)in the inoculating sludge were preserved,many bands disappeared or weakened (bands 3,4,6,8,13,14,15).After day 45,the diversity index tended to be stable and showed small fluctuation (from 0.72to 0.82).Banding pattern similarity was analyzed by applying UPGMA (Fig.4)algorithms.The UPGMA analysis showed three groups with intragroup similarity at approximately 67%e 78%and intergroup similarity at 44e 62%.Generally,the clustering followed the time course;and the algorithms showed a closer clustering of groups II and III.In the analysis,group I was associated with sludge inoculation and washout,group IIwithFig.3e DGGE profile of the AOB communities in the SBR during the sludge granulation process (lane labels along the top show the sampling time (days)from startup of the bioreactor).The major bands were labeled with the numbers (bands 1e15).Fig.4e UPGMA analysis dendrograms of AOB community DGGE banding patterns,showing schematics of banding patterns.Roman numerals indicate major clusters.w a t e r r e s e a r c h x x x (2011)1e 105startup sludge granulation and decreasing SVI 10,and group III with a stable system and excellent biomass settleability.In Fig.3,the locations of the predominant bands were excised from the gel.DNA in these bands were reamplified,cloned and sequenced.The comparative analysis of these partial 16S rRNA sequences (Table 2and Fig.S6)revealed the phylogenetic affiliation of 13sequences retrieved.The majority of the bacteria in seed sludge grouped with members of Nitrosomonas and Nitrosospira .Along with sludge granula-tion,most of Nitrosomonas (Bands 2,5,7,9,10,11)were remained or eventually became dominant in GSBR;however,all of Nitrosospira (Bands 6,13,15)were gradually eliminated from the reactor.3.3.Distribution of AOB and NOB in different sized granulesFISH was performed on the granule sections mainly to deter-mine the location of AOB and NOB within the different size classes of granules,and the images were not further analyzed for quantification of cell counts.As shown in Fig.6,in small granules (0.2mm <d <0.45mm),AOB located mainly in the outer part of granular space,whereas NOB were detected only in the core of granules.In medium granules (0.45mm <d <0.6mm),AOB distributed evenly throughout the whole granular space,whereas NOB still existed in the inner part.In the larger granules (d >0.9mm),AOB and NOB were mostly located in the surface area of the granules,and moreover,NOB became rare.4.Discussion4.1.Relationship between granule formation and reactor performanceAfter day 32,the SVI 10stabilized at 20e 35mL g À1,which is very low compared to the values measured for activated sludge (100e 150mL g À1).However,the size distribution of the granules measured on day 32(Fig.2)indicated that only 22%of the biomass was made of granular sludge with diameter largerthan 0.2mm.These results suggest that sludge settleability increased prior to granule formation and was not affected by different particle sizes in the sludge during the GSBR operation.It was observed,however,that the diameter of the granules fluctuated over longer durations.The large granules tended to destabilize due to endogenous respiration,and broke into smaller granules that could seed the formation of large granules again.Pochana and Keller reported that physically broken sludge flocs contribute to lower denitrification rates,due to their reduced anoxic zone (Pochana and Keller,1999).Therefore,TN removal efficiency raises fluctuantly throughout the experiment.Some previous research had demonstrated that bigger,more dense granules favored the enrichment of PAO (Winkler et al.,2011a ).Hence,after day 77,removal efficiency of TP was higher and relatively stable because the granules mass fraction was over 90%and more larger granules formed.4.2.Relationship between AOB communities dynamic and sludge granulationFor granule formation,a short settling time was set,and only particles with a settling velocity higher than 4.5m h À1were retained in the reactor.Moreover,as shown in Fig.1,the variation in SVI 10was greater before day 41(from 108.2mL g À1e 34.1mL g À1).During this phase,large amounts of biomass could not survive in the reactor.A clear shift in pop-ulations was evident,with 58%similarity between days 8and 18(Table S1).In the SBR system fed with acetate-based synthetic wastewater,heterotrophic bacteria can produce much larger amounts of extracellular polysaccharides than autotrophic bacteria (Tsuneda et al.,2003).Some researchers found that microorganisms in high shear environments adhered by extracellular polymeric substances (EPS)to resist the damage of suspended cells by environmental forces (Trinet et al.,1991).Additionally,it had been proved that the dominant heterotrophic species in the inoculating sludge were preserved throughout the process in our previous research (Zhang et al.,2011).It is well known that AOB are chemoau-totrophic and slow-growing;accordingly,numerous AOBw a t e r r e s e a r c h x x x (2011)1e 106populations that cannot become big and dense enough to settle fast were washed out from the system.As a result,the variation in AOB was remarkable in the period of sludge inoculation,and the diversity index of population decreased rapidly.After day 45,AOB communities’structure became stable due to the improvement of sludge settleability and the retention of more biomass.These results suggest that the short settling time (selection pressure)apparently stressed the biomass,leading to a violent dynamic of AOB communities.Further,these results suggest that certain populations may have been responsible for the operational success of the GSBR and were able to persist despite the large fluctuations in pop-ulation similarity.This bacterial population instability,coupled with a generally acceptable bioreactor performance,is congruent with the results obtained from a membrane biore-actor (MBR)for graywater treatment (Stamper et al.,2003).Nitrosomonas e like and Nitrosospira e like populations are the dominant AOB populations in wastewater treatment systems (Kowalchuk and Stephen,2001).A few previous studies revealed that the predominant populations in AOB communities are different in various wastewater treatment processes (Tawan et al.,2005;Thomas et al.,2010).Some researchers found that the community was dominated by AOB from the genus Nitrosospira in MBRs (Zhang et al.,2010),whereas Nitrosomonas sp.is the predominant population in biofilter sludge (Yin and Xu,2009).In the currentstudy,Fig.5e DGGE profile of the AOB communities in different size of granules (lane labels along the top show the range of particle diameter (d,mm)).Values along the bottom indicate the Shannon diversity index (H ).Bands labeled with the numbers were consistent with the bands in Fig.3.w a t e r r e s e a r c h x x x (2011)1e 107sequence analysis revealed that selection pressure evidently effect on the survival of Nitrosospira in granular sludge.Almost all of Nitrosospira were washed out initially and had no chance to evolve with the environmental changes.However,some members of Nitrosomonas sp.have been shown to produce more amounts of EPS than Nitrosospira ,especially under limited ammonia conditions (Stehr et al.,1995);and this feature has also been observed for other members of the same lineage.Accordingly,these EPS are helpful to communicate cells with each other and granulate sludge (Adav et al.,2008).Therefore,most of Nitrosomonas could adapt to this challenge (to become big and dense enough to settle fast)and were retained in the reactor.At the end of reactor operation (day 180),granules with different particle size were sieved.The effects of variation in granules size on the composition of the AOBcommunitiesFig.6e Micrographs of FISH performed on three size classes of granule sections.DAPI stain micrographs (A,D,G);AOB appear as green fluorescence (B,E,H),and NOB appear as red fluorescence (C,F,I).Bar [100m m in (A)e (C)and (G)e (I).d,particle diameter.(For interpretation of the references to colour in this figure legend,the reader is referred to the web version of this article.)w a t e r r e s e a r c h x x x (2011)1e 108were investigated.As shown in Fig.5,AOB communities structures in different size of granules were varied.Although several predominant bands(bands2,5,11)were present in all samples,only bands3and6appeared in the granules with diameters larger than0.6mm.Additionally,bands7and10 were intense in the granules larger than0.45mm.According to Table2,it can be clearly indicated that Nitrosospira could be retained merely in the granules larger than0.6mm.Therefore, Nitrosospira was not present at a high level in Fig.3due to the lower proportion of larger granules(d>0.6mm)in TSS along with reactor operation.DGGE analysis also revealed that larger granules had a greater microbial diversity than smaller ones. This result also demonstrates that more organisms can survive in larger granules as a result of more space,which can provide the suitable environment for the growth of microbes(Fig.6).4.3.Effect of variance in particle size on the distribution of AOB and NOB in granulesAlthough an influence of granule size has been observed in experiments and simulations for simultaneous N-and P-removal(de Kreuk et al.,2007),the effect of granule size on the distribution of different biomass species need be revealed further with the assistance of visible experimental results, especially in the same granular sludge reactors.Related studies on the diversity of bacterial communities in granular sludge often focus on the distribution of important functional bacteria populations in single-size granules(Matsumoto et al., 2010).In the present study,different size granules were sieved,and the distribution patterns of AOB and NOB were explored.In the nitrification processes considered,AOB and NOB compete for space and oxygen in the granules(Volcke et al.,2010).Since ammonium oxidizers have a higheroxygen affinity(K AOBO2<K NOBO2)and accumulate more rapidly inthe reactor than nitrite oxidizers(Volcke et al.,2010),NOB are located just below the layer of AOB,where still some oxygen is present and allows ready access to the nitrite produced.In smaller granules,the location boundaries of the both biomass species were distinct due to the limited existence space provided by granules for both microorganism’s growth.AOB exist outside of the granules where oxygen and ammonia are present.Medium granules can provide broader space for microbe multiplying;accordingly,AOB spread out in the whole granules.This result also confirms that oxygen could penetrate deep into the granule’s core without restriction when particle diameter is less than0.6mm.Some mathematic model also supposed that NOBs are favored to grow in smaller granules because of the higher fractional aerobic volume (Volcke et al.,2010).As shown in the results of the batch experiments(Zhang et al.,2011),nitrite accumulation temporarily occurred,accompanied by the more large gran-ules(d>0.9mm)forming.This phenomenon can be attrib-uted to the increased ammonium surface load associated with larger granules and smaller aerobic volume fraction,resulting in outcompetes of NOB.It also suggests that the core areas of large granules(d>0.9mm)could provide anoxic environment for the growth of anaerobic denitrificans(such as Tb.deni-trificans or Tb.thioparus in Fig.S7,Supporting information).As shown in Fig.2and Fig.S3,the removal efficiency of total nitrogen increased with formation of larger granules.5.ConclusionsThe variation in AOB communities’structure was remarkable during sludge inoculation,and the diversity index of pop-ulation decreased rapidly.Most of Nitrosomonas in the inocu-lating sludge were retained because of their capability to rapidly adapt to the settling e washing out action.DGGE anal-ysis also revealed that larger granules had greater AOB diversity than that of smaller ones.Oxygen penetration was not restricted in the granules of less than0.6mm particle diameter.However,the larger granules(d>0.9mm)can result in the smaller aerobic volume fraction and inhibition of NOB growth.Henceforth,further studies on controlling and opti-mizing distribution of granule size could be beneficial to the nitrogen removal and expansive application of granular sludge technology.AcknowledgmentsThis work was supported by grants from the National Natural Science Foundation of China(No.51108456,50908227)and the National High Technology Research and Development Program of China(No.2009AA06Z312).Appendix.Supplementary dataSupplementary data associated with this article can be found in online version at doi:10.1016/j.watres.2011.09.026.r e f e r e n c e sAdav,S.S.,Lee, D.J.,Show,K.Y.,2008.Aerobic granular sludge:recent advances.Biotechnology Advances26,411e423.APHA-AWWA-WEF,2005.Standard Methods for the Examination of Water and Wastewater,first ed.American Public Health Association/American Water Works Association/WaterEnvironment Federation,Washington,DC.de Bruin,L.M.,de Kreuk,M.,van der Roest,H.F.,Uijterlinde,C., van Loosdrecht,M.C.M.,2004.Aerobic granular sludgetechnology:an alternative to activated sludge?Water Science and Technology49,1e7.de Kreuk,M.,Heijnen,J.J.,van Loosdrecht,M.C.M.,2005.Simultaneous COD,nitrogen,and phosphate removal byaerobic granular sludge.Biotechnology and Bioengineering90, 761e769.de Kreuk,M.,Picioreanu,C.,Hosseini,M.,Xavier,J.B.,van Loosdrecht,M.C.M.,2007.Kinetic model of a granular sludge SBR:influences on nutrient removal.Biotechnology andBioengineering97,801e815.Downing,L.S.,Nerenberg,R.,2008.Total nitrogen removal ina hybrid,membrane-aerated activated sludge process.WaterResearch42,3697e3708.Erguder,T.H.,Boon,N.,Vlaeminck,S.E.,Verstraete,W.,2008.Partial nitrification achieved by pulse sulfide doses ina sequential batch reactor.Environmental Science andTechnology42,8715e8720.w a t e r r e s e a r c h x x x(2011)1e109。

植物[编辑]物种基因组大小和开放阅读框文献Sesamum indicum L. Sesame 芝麻(2n = 26)293.7 Mb, 10,656 orfs 1 Oryza brachyantha短药野生稻261 Mb, 32,038 orfs 2Chondrus crispus Red seaweed爱尔兰海藻105 Mb, 9,606 orfs 3Pyropia yezoensis susabi-nori海苔43 Mb, 10,327 orfs 4Prunus persica Peach 桃226.6 of 265 Mb 27,852 orfs 5Aegilops tauschii 山羊草(DD)4.23 Gb (97% of the 4.36), 43,150 orfs 6 Triticum urartu 乌拉尔图小麦(AA)4.66 Gb (94.3 % of 4.94 Gb, 34,879 orfs 7 moso bamboo (Phyllostachys heterocycla) 毛竹2.05 Gb (95%) 31,987 orfs 8 Cicer arietinum Chickpea鹰嘴豆~738-Mb,28,269 orfs 9 520 Mb (70% of 740 Mb), 27,571 orfs 10Prunus mume 梅280 Mb, 31,390 orfs 11Gossypium hirsutum L.陆地棉2.425 Gb 12Gossypium hirsutum L. 雷蒙德氏棉761.8 Mb 13Citrus sinensis 甜橙87.3% of ~367 Mb, 29,445 orfs 14甜橙367 Mb 15Citrullus lanatus watermelon 西瓜353.5 of ~425 Mb (83.2%) 23,440 orfs 16 Betula nana dwarf birch,矮桦450 Mb 17Nannochloropsis oceanica CCMP1779微绿球藻(产油藻类之一)28.7 Mb,11,973 orfs 18Triticum aestivum bread wheat普通小麦17 Gb, 94,000 and 96,000 orfs 19 Hordeum vulgare L. barley 大麦1.13 Gb of 5.1 Gb,26,159 high confidence orfs,53,000 low confidence orfs 20Gossypium raimondii cotton 雷蒙德氏棉D subgenome,88% of 880 Mb 40,976 orfs 21Linum usitatissimum flax 亚麻302 mb (81%), 43,384 orfs 22Musa acuminata banana 香蕉472.2 of 523 Mb, 36,542 orfs 23Cucumis melo L. melon 甜瓜375 Mb(83.3%)27,427 orfs 24Pyrus bretschneideri Rehd. cv. Dangshansuli 梨(砀山酥梨)512.0 Mb (97.1%), 42,812 orfs 25,26Solanum lycopersicum 番茄760/900 Mb,34727 orfs 27S. pimpinellifolium LA1589野生番茄739 MbSetaria 狗尾草属(谷子、青狗尾草)400 Mb,25000-29000 orfs 28,29Cajanus cajan pigeonpea木豆833 Mb,48,680 orfs 30Nannochloropis gaditana 一种海藻~29 Mb, 9,052 orfs 31Medicago truncatula蒺藜苜蓿350.2 Mb, 62,388 orfs 32Brassica rapa 白菜485 Mb 33Solanum tuberosum 马铃薯0.73 Mb,39031 orfs 34Thellungiella parvula条叶蓝芥13.08 Mb 29,338 orfs 35Arabidopsis lyrata lyrata 玉山筷子芥? 183.7 Mb, 32670 orfs 36Fragaria vesca 野草莓240 Mb,34,809 orfs 37Theobroma cacao 可可76% of 430 Mb, 28,798 orfs 38Aureococcus anophagefferens褐潮藻32 Mb, 11501 orfs 39Selaginella moellendorfii江南卷柏208.5 Mb, 34782 orfs 40Jatropha curcas Palawan麻疯树285.9 Mb, 40929 orfs 41Oryza glaberrima 光稃稻(非洲栽培稻)206.3 Mb (0.6x), 10 080 orfs (>70% coverage) 42Phoenix dactylifera 棕枣380 Mb of 658 Mb, 25,059 orfs 43Chlorella sp. NC64A小球藻属40000 Kb, 9791 orfs 44Ricinus communis蓖麻325 Mb, 31,237 orfs 45Malus domestica (Malus x domestica) 苹果742.3 Mb 46Volvox carteri f. nagariensis 69-1b一种团藻120 Mb, 14437 orfs 47Brachypodium distachyon 短柄草272 Mb,25,532 orfs 48Glycine max cultivar Williams 82栽培大豆1.1 Gb, 46430 orfs 49Zea mays ssp. Mays Zea mays ssp. Parviglumis Zea mays ssp. Mexicana Tripsacum dactyloides var. meridionale 无法下载附表50Zea mays mays cv. B73玉米2.06 Gb, 106046 orfs 51Cucumis sativus 9930 黄瓜243.5 Mb, 63312 orfs 52Micromonas pusilla金藻21.7 Mb, 10248 orfs 53Sorghum bicolor 高粱697.6 Mb, 32886 orfs 54Phaeodactylum tricornutum 三角褐指藻24.6 Mb, 9479 orfs 55Carica papaya L. papaya 番木瓜271 Mb (75%), 28,629 orfs 56Physcomitrella patens patens小立碗藓454 Mb, 35805 orfs 57Vitis vinifera L. Pinot Noir, clone ENTAV 115葡萄504.6 Mb, 29585 orfs 58Vitis vinifera PN40024葡萄475 Mb 59Ostreococcus lucimarinus绿色鞭毛藻13.2 Mb, 7640 orfs 60Chlamydomonas reinhardtii 莱茵衣藻100 Mb, 15256 orfs 61Populus trichocarpa黑三角叶杨550 Mb, 45000 orfs 62Ostreococcus tauri 绿藻12.6 Mb, 7892 orfs 63Oryza sativa ssp. japonica 粳稻360.8 Mb, 37544 orfs 64Thalassiosira pseudonana 硅藻25 Mb, 11242 orfs 65Cyanidioschyzon merolae 10D红藻16.5 Mb, 5331 orfs 66Oryza sativa ssp. japonica 粳稻420 Mb, 50000 orfs 67Oryza sativa L. ssp. Indica籼稻420 Mb, 59855 orfs 68Guillardia theta -蓝隐藻,551 Kb, 553 orfs 69Arabidopsis thaliana Columbia拟南芥119.7 Mb, 31392 orfs 70参考文献1 Zhang, H. et al. Genome sequencing of the important oilseed crop Sesamum indicum L. Genome Biology 14, 401 (2013).2 Chen, J. et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nat Commun 4, 1595 (2013).3 Collén, J. et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proceedings of the National Academy of Sciences 110, 5247-5252 (2013).4 Nakamura, Y. et al. The first symbiont-free genome sequence of marine red alga, susabi-nori Pyropia yezoensis. PLoS ONE 8, e57122 (2013).5 Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genetics advance online publication (2013).6 Jia, J. et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91-95 (2013).7 Ling, H.-Q. et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496, 87-90 (2013).8 Peng, Z. et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nature Genetics 45, 456-461 (2013).9 Jain, M. et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant Journal, DOI: 10.1111/tpj.12173 (2013).10 Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotech 31, 240-246 (2013).11 Zhang, Q. et al. The genome of Prunus mume. Nat Commun 3, 1318 (2012).12 Lee, M.-K. et al. Construction of a plant-transformation-competent BIBAC library and genome sequence analysis of polyploid Upland cotton (Gossypium hirsutum L.). BMC Genomics 14, 208 (2013).13 Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423-427 (2012).14 Xu, Q. et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet 45, 59–66 (2013).15 Belknap, W. R. et al. Characterizing the citrus cultivar Carrizo genome through 454 shotgun sequencing. Genome 54, 1005-1015 (2011).16 Guo, S. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45, 51–58 (2013).17 Wang, N. et al. Genome sequence of dwarf birch (Betula nana) andcross-species RAD markers. Mol Ecol Article first published online: 21 NOV 2012 DOI: 10.1111/mec.12131 (2012).18 Vieler, A. et al. Genome, functional gene annotation, and nuclear transformation of the heterokont oleaginous alga Nannochloropsis oceanica CCMP1779. PLoS Genet 8, e1003064 (2012).19 Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705-710 (2012).20 Consortium, T. I. B. G. S. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012).21 Wang, K. et al. The draft genome of a diploid cotton Gossypium raimondii. Nature Genetics 44, 1098–1103 (2012).22 Wang, Z. et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. The Plant Journal 72, 461-473 (2012).23 D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).24 Garcia-Mas, J. et al. The genome of melon (Cucumis melo L.). PNAS 109, 11872-11877 (2012).25 reporter, A. G. s. Consortium releases pear genome data. GenomeWeb Daily News (2012).26 Wu, J. et al. The genome of pear (Pyrus bretschneideri Rehd.). Genome Res.Published in Advance November 13, 2012, doi:10.1101/gr.144311.112 (2012).27 Consortium, T. T. G. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).28 Bennetzen, J. L. et al. Reference genome sequence of the model plant Setaria. Nat Biotech 30, 555-561 (2012).29 Zhang, G. et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotech 30, 549-554 (2012).30 Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotech 30, 83-89 (2012).31 Radakovits, R. et al. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana. Nat Commun 3, 686 (2012).32 Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).33 Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035-1039 (2011).34 Consortium, T. P. G. S. Genome sequence and analysis of the tuber crop potato. Nature 475, 189-195 (2011).35 Dassanayake, M. et al. The genome of the extremophile crucifer Thellungiella parvula. Nat. Genet. 43, 913-918 (2011).36 Hu, T. T. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476-481 (2011).37 Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109-116 (2011).38 Argout, X. et al. The genome of Theobroma cacao. Nat. Genet. 43, 101-108 (2011).39 Gobler, C. J. et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. PNAS 108, 4352-4357 (2011).40 Banks, J. A. et al. The selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332, 960-963 (2011).41 Sato, S. et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 18, 65-76 (2011).42 Sakai, H. et al. Distinct evolutionary patterns of Oryza glaberrima deciphered by genome sequencing and comparative analysis. Plant Journal 66, 796-805 (2011).43 Al-Dous, E. K. et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotech 29, 521-527 (2011).44 Blanc, G. et al. The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell 22,2943-2955 (2010).45 Chan, A. P. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotech 28(951-956 (2010).46 Velasco, R. et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 42, 833-839 (2010).47 Prochnik, S. E. et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 329, 223-226 (2010).48 Initiative, T. I. B. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763-768 (2010).49 Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178-183 (2010).50 Hufford, M. B. et al. Comparative population genomics of maize domestication and improvement. Nat Genet 44, 808-811 (2012).51 Wei, F. et al. The physical and genetic framework of the maize B73 genome. PLoS Genet 5, e1000715 (2009).52 Huang, S. et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275-1281 (2009).53 Worden, A. Z. et al. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324, 268-272 (2009).54 Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551-556 (2009).55 Bowler, C. et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456, 239-244 (2008).56 Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991-996 (2008).57 Rensing, S. A. et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319, 64-69 (2008).58 Velasco, R. et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2, e1326 (2007).59 Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463-467 (2007).60 Palenik, B. et al. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. PNAS 104, 7705-7710 (2007).61 Merchant, S. S. et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318, 245-250 (2007).62 Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596-1604 (2006).63 Derelle, E. et al. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. PNAS 103, 11647-11652 (2006).64 Project, I. R. G. S. The map-based sequence of the rice genome. Nature 436, 793-800 (2005).65 Armbrust, E. V. et al. The genome of the diatom Thalassiosira Pseudonana: ecology, evolution, and metabolism. Science 306, 79-86 (2004).66 Matsuzaki, M. et al. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428, 653-657 (2004).67 Goff, S. A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92-100 (2002).68 Yu, J. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296, 79-92 (2002).69 Douglas, S. et al. The highly reduced genome of an enslaved algal nucleus. Nature 410, 1091-1096 (2001).70 Kaul, S. et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796-815 (2000).动物[编辑]•Anopheles gambiae - 疟蚊•Apis mellifera - 蜜蜂•Bos taurus cattle - 牛•Caenorhabditis briggsae - 一种线虫•Caenorhabditis elegans - 秀丽隐杆线虫,模式生物•Canis lupus familiaris dog- 狗•Ciona intestinalis - 一种海鞘•Ciona savignyi - 一种海鞘•Drosophila melanogaster - 黑腹果蝇,模式生物•Fugu rubripes - 河豚•Gallus gallus - 鸡•Homo sapiens - 人•Mus musculus - 小鼠, 模式生物•Pan troglodytes - 黑猩猩•Rattus norvegicus - 大鼠•Schistosoma haematobium - 埃及血吸虫•Anabaena spec. - Fadenblaualge•Gloeobacter violaceus - primitive Blaualge •Synechococcus spec. - Meeres-Blaualge•Synechocystis spec. - Meeres-Blaualge•Thermosynechococcus elongatus - Thermophile Blaualge。

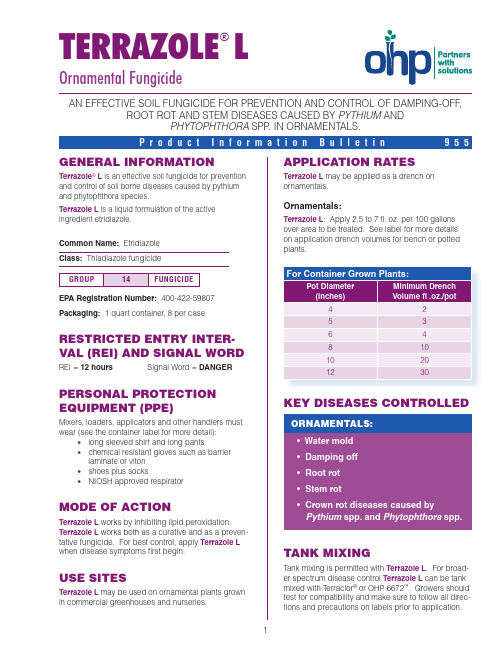
GENERAL INFORMATIONTerrazole ® L is an effective soil fungicide for prevention and control of soil borne diseases caused by pythium and phytophthora species.Terrazole L is a liquid formulation of the active ingredient etridiazole.Common Name: Etridiazole Class: Thiadiazole fungicideEPA Registration Number: 400-422-59807Packaging: 1 quart container, 8 per caseRESTRICTED ENTRY INTER-VAL (REI) AND SIGNAL WORDREi = 12 hourssignal Word = DANGERPERSONAL PROTECTION EQUIPMENT (PPE)Mixers, loaders, applicators and other handlers must wear (see the container label for more detail):• long sleeved shirt and long pants• chemical resistant gloves such as barrier laminate or viton • shoes plus socks• NiosH approved respiratorMODE OF ACTIONTerrazole L works by inhibiting lipid peroxidation. Terrazole L works both as a curative and as a preven-tative fungicide. For best control, apply Terrazole L when disease symptoms first begin.USE SITESTerrazole L may be used on ornamental plants grown in commercial greenhouses and nurseries.APPLICATION RATESTerrazole L may be applied as a drench on ornamentals.Ornamentals:Terrazole L : Apply 2.5 to 7 fl. oz. per 100 gallons over area to be treated. see label for more details on application drench volumes for bench or potted plants.:KEY DISEASES CONTROLLEDTANK MIXINGTank mixing is permitted with Terrazole L . For broad-er spectrum disease control Terrazole L can be tank mixed with Terraclor ® or oHp 6672™. Growers should test for compatibility and make sure to follow all direc-tions and precautions on labels prior to application.1For Container Grown Plants:Pot Diameter (inches)Minimum Drench Volume fl .oz./pot4 25 3648 1010 201230Terrazole and Terraclor are registered trademarks of Chemtura Corp. Aliette is a registered trademark and FenStop is a trademark of Bayer. Disarm is a trademark of Arysta LifeScience North America, LLC. oHp 6672 is a trademark of oHp , inc.© oHp , inc. 03/20102oHp955 03/10po Box 230, Mainland, pA 19451Technical service: use of Terrazole L according to the labeling is subject to the use precautions and limitations imposed by the label affixed to the container for Terrazole L. it is a violation of Federal law to use this product in a manner inconsistent with its labeling.4Disarm。
![组成型植物启动子[发明专利]](https://img.taocdn.com/s1/m/31def20319e8b8f67d1cb945.png)
专利名称:组成型植物启动子
专利类型:发明专利
发明人:M·T·J·德鲍思,N·E·M·克瓦埃德福里格,B·G·J·费尔兰斯-昂斯腾克,L·K·瓦乔斯基
申请号:CN200680052639.8
申请日:20061212
公开号:CN101370939A
公开日:
20090218
专利内容由知识产权出版社提供
摘要:提供了在本文中称为AA6启动子的强组成型植物启动子,该启动子在生物和/或非生物胁迫条件下保持强组成型活性。
还提供了含有AA6启动子的转基因细胞和生物体,具体是植物细胞和植物,以及使用AA6启动子在细胞和生物体中表达核酸序列的方法。
申请人:关键基因公司
地址:荷兰瓦格宁根
国籍:NL
代理机构:北京北翔知识产权代理有限公司
更多信息请下载全文后查看。

【人物与科研】自然资源部第三海洋...珊瑚礁是世界上生物生产力最高的生态系统之一,研究人员从中发现了大量具有新结构和多种生物活性的天然产物。
健康珊瑚的粘液层、骨骼和组织通常含有大量的真核藻类、细菌和古细菌。
这些微生物产生的多种多样的生物活性次级代谢产物有助于珊瑚宿主更好地面对捕食者和激烈的竞争。
近日,自然资源部第三海洋研究所杨献文研究员课题组从珊瑚来源的放线菌Nesterenkonia halobia(N. halobia)中分离得到一种罕见的笼状聚酮类天然产物nesteretal A(图1),并通过多种光谱学手段和计算方法确定了其结构(Organic Letters, 2019, DOI: 10.1021/lett.9b02634)。
图1 珊瑚来源的放线菌N. halobia及其次级代谢产物nesteretal A(来源:Organic Letters)杨献文,自然资源部第三海洋研究所研究员,博士生导师。
1998年本科毕业于沈阳药科大学,2006年博士毕业于中国科学院昆明植物研究所,2006年-2008年在上海第二军医大学开展博士后研究,2008年-2015年在中国科学院南海海洋研究所任副研究员,2011年-2015年在卢森堡健康研究院任研究员。
2015年加入自然资源部第三海洋研究所,研究方向为海洋微生物天然药物化学研究。
杨献文研究员先后主持了20多项国家或省部级等科研项目,已分离鉴定化合物3000多个,其中新结构300多个;相关研究成果已发表SCI学术论文150多篇,其中以第一作者或通讯作者身份在Chem. Commun., Org. Lett.等刊物上发表论文60多篇;已授权或申请国内及国际专利20多项;现为Frontiers in Chemistry的客座副主编,同时也是Org. Lett., J. Nat. Prod.等多个国际期刊的审稿人。
来自海洋放线菌N. halobia的新颖笼状聚酮化合物海洋放线菌是活性天然产物的重要来源,前期,课题组已经从深海放线菌Nesterenkonia flava(N. flava)中分离鉴定了13个化合物。

哈佛大学新研究揭示海绵基因组传达遗传复杂性的出现哈佛大学最近研究了四个纲八个种海绵的转录组,专门寻找与动物复杂性相关的基因和途径,成果发表在Mol Biol Evol上。
哈佛大学最近研究了四个纲(Hexactinellida, Demospongiae, Homoscleromorpha and Calcarea)八个种海绵的转录组,专门寻找与动物复杂性相关的基因和途径,成果发表在Mol Biol Evol上。
海绵(多孔动物)是最早进化的动物,它滤食性身体计划是由复杂的含水系统组成的环细胞室组成的,在后生动物中非常独特。
它表示海绵与其他动物在肌肉和神经功能进化之前早有分歧,或表示海绵已失去这些特征。
Amphimedon和Oscarella基因组的分析支持这一观点——许多后生动物的关键基因在所研究的海绵中的是不存在的,但其他海绵中这些基因的存在是未知的。
哈佛大学最近研究了四个纲(Hexactinellida, Demospongiae, Homoscleromorpha and Calcarea)八个种海绵的转录组,专门寻找与动物复杂性相关的基因和途径,成果发表在Mol Biol Evol上。
他们在三种单细胞后鞭毛生物和两种两侧对称动物类群的转录组和基因组中寻找这些基因作为参考。
他们的分析表明,所有海绵纲与其他后生动物共享补充基因。
该团队发现Hexactinellid, Calcareous and Homoscleromorph三种海绵与非两侧对称动物相比共享给两侧对称动物更多的基因(由联川生物提供poly(A)RNA测序服务)。
他们还发现大多数分子代表参与细胞与细胞间的通信,发出信号,活跃在复杂的上皮细胞中,免疫识别和生殖系/性别,只有少数潜在的关键分子没有参与。
一个值得注意的发现是,所有寻常海绵纲(转录组和Amphimedon基因组)某些重要基因的缺失可能反映了主干谱系包括Hexactinellid, Calcareous and Homoscleromorph的分歧。

An Exhaustive Genome Assembly Algorithm Using K-Mers to IndirectlyPerform N-Squared Comparisons in O(N)Maulik K. Shah 1,2, HoJoon Lee 1, Stephanie A. Rogers 1, Jeffrey W. Touchman 2,31Computational Biosciences Program, Arizona State University, Tempe, Arizona2Translational Genomics Research Institute, Phoenix, Arizona 3School of Life Sciences, Arizona State University, Tempe, ArizonaEmail: maulik.k.shah@AbstractWe pre se nt an algorithm that indire ctly make s N 2sequence comparisons in O(N) with respect to the size of the ge nome. This algorithm is ve ry applicable inasse mbling whole ge nome s from thethousands of DNA s e qu e nc e fragm e nts that ar e g e n e rat e d in shotgun se que ncing. First, we assume that fragme nts that shar e k-m e rs should ov e rlap in th efinal assembly. We then catalog all k-mers that exist in the shotgun library and infer links between fragments that share k-me rs. The se links are the n use d to re pre se nt edges in a graph. This graph is generated in O(N) yet re pre se nts the re sult of comparing e ve ry fragme nt to every other fragment.1. IntroductionThe complete sequence of an organism’s genome is an incredibly valuable tool for biological research. Using today’s technology, the most common method of elucidating the sequence is shotgun sequencing followed by in-silico assembly.An example description of sequencing a large microbial genome is as follows. Approximately five million base pairs are assembled from 70,000 sequence fragments, where each is approximately 800 base pairs in length. The most straightforward approach would be to compare each fragment to all others, identify fragments that overlap, and then rebuild the original sequence. However, the time to directly make all comparisons will grow exponentially with the size of the genome. We have developed an algorithm to indirectly make all comparisons in linear running time. Because our approach effectively makes all of these comparisons and does not abandon the traditional ‘overlap-layout-consensus’ approach, we feel it will make more accurate assemblies than non-exhaustive approaches such as those used in ARACHNE [1], PHRAP [2],and EULER [3].2. Methods2.1. K-Mer LibraryTo make all N 2 comparisons, we begin by building a library of k-mers (sequences that consist of nucleotides of length k). Scanning through each fragment in the dataset by using a sliding window of size k, we catalog each of these k-mers and the fragment in which it occurred. Since we assume that fragments that neighbored each other in the complete genomic sequence would share unique k-mers, cataloging the k-mers in this fashion would indirectly identify neighboring fragments. Unlike ARACHNE , we do not attempt to minimize memory requirements, but rather evaluate all k-mers.2.2. Adjacency TableThe k-mer library shows which fragments share k-mers. However, because we used a sliding window, a pair of neighboring fragments would share as many k-mers as the length of their overlap. However, the only relevant information is that these two fragments overlap by at least k base pairs and therefore belong next to one another in the complete genome. Therefore, we built a new table that contained only one link per pair of overlapping fragments, no matter the number of k-mers shared. This table is particularly significant as it represents all N 2 comparisons and was generated in O(N)2.3. Identifying ContigsThe adjacency table is the most valuable data structure in this algorithm. We viewed the adjacencytable as a set of disjoint, undirected, cyclical graphs; a similar approach to EULER. Identifying and separating these graphs would result in a representation of smaller segments of the genome. Traditional assemblers call these segments contigs and each disjoint graph would represent a single contig. We employed multiple breadth first searches to identify these disjoint graphs. Since this process explores each fragment only once, it also runs in linear time.3. ResultsTwo datasets were used in this experiment. The first was a simulated shotgun library derived from the complete genome of the CO92 strain of Y.Pestis. We artificially generated a shotgun library containing 41,400 random fragments which represents nine-fold coverage. We also used a real shotgun library of the FV-1 strain of Y.Pestis which contained 70,176 fragments. Using 24 as the size of k, the CO92 dataset showed that 39,809 of the reads were linked into a single graph and the FV-1 dataset showed that 41,539 fragments were linked into a single graph. We had initially hypothesized that this approach would yield similar results to ARACHNE and PHRAP. However, it seems to have connected more fragments than expected.The following figures were generated by running the algorithm on data from the FV-1 Y.Pestis. shotgun library. Figure 1 shows how the occurrence of k-merswere distributed.Figure 1 – K-mer distributionNote that Figure 2 shows that on average, a node in the graph had 9.5 neighbors. This is consistent with our expectations as the shotgun library had over nine-fold coverage.Figure 2 – Connectivity of the nodes in the graph 4. ConclusionsBy combining concepts found in both ARACHNE and EULER, we plan to unite these methods of usingoverlap-layout-consensus with building graphs to assemble a genome with higher accuracy. Although we have yet to make use of mate-pair information, error-correction or repeat-masking to help in finding the solution, initial results are promising.We are grateful to Steve Mastrian, Debbie Benitez, Erin Clark, Jicheng Hao, Dave Youngkin, Andrzej Czygrinow and Rosemary Renaut for providing guidance and sequencing data.5. References[1] Batzoglou, S., Jaffe, D., Stanley, K., Butler, J., Gnerre, S., Mauceli, E., Berger, B., Mesirov, J.P., and Lander, E.S. ARACHNE: A whole-genome shotgun assembler. Genome Res. 2002 Jan 1; 12(1): 177–189.[2] Green P. (1994) Documentation for Phrap (/phrap.docs/phrap.html)[3] Pevzner PA, Tang H, Waterman M S. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci USA. 2001 Aug 14; 98(17): 9748-9753。