拟肾上腺素药
- 格式:pptx
- 大小:505.09 KB
- 文档页数:43





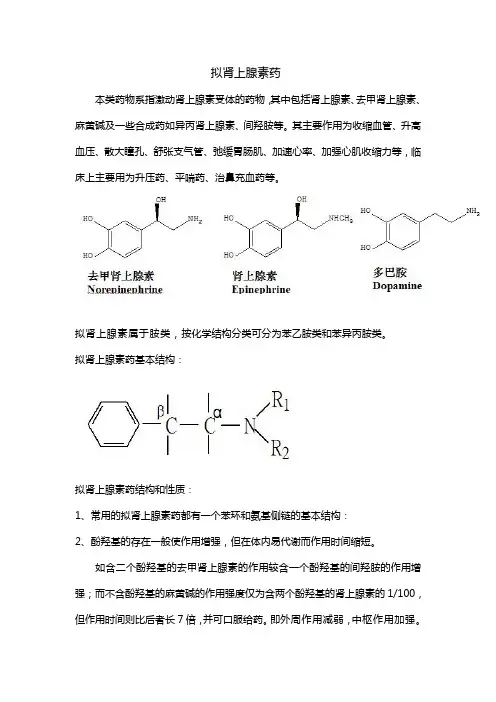
拟肾上腺素药本类药物系指激动肾上腺素受体的药物,其中包括肾上腺素、去甲肾上腺素、麻黄碱及一些合成药如异丙肾上腺素、间羟胺等。
其主要作用为收缩血管、升高血压、散大瞳孔、舒张支气管、弛缓胃肠肌、加速心率、加强心肌收缩力等,临床上主要用为升压药、平喘药、治鼻充血药等。
拟肾上腺素属于胺类,按化学结构分类可分为苯乙胺类和苯异丙胺类。
拟肾上腺素药基本结构:拟肾上腺素药结构和性质:1、常用的拟肾上腺素药都有一个苯环和氨基侧链的基本结构:2、酚羟基的存在一般使作用增强,但在体内易代谢而作用时间缩短。
如含二个酚羟基的去甲肾上腺素的作用较含一个酚羟基的间羟胺的作用增强;而不含酚羟基的麻黄碱的作用强度仅为含两个酚羟基的肾上腺素的1/100,但作用时间则比后者长7倍,并可口服给药。
即外周作用减弱,中枢作用加强。
3、氨基侧链的β-碳上的醇羟基对活性的影响表现在立体异构体间的差别。
左旋体手性β-碳原子都是R构型,其活性明显高于右旋体,β受体效应尤为显著。
如异丙肾上腺素左旋体的β2受体效应(支气管扩张作用)比右旋体强800倍;再如麻黄碱四个光学异构体中只有(-)(1R,2S)-麻黄碱的活性最强,其具有醇羟基的碳原子是R构型。
4、氨基侧链的α-碳上引入甲基时,则肾上腺素受体激动作用减弱,中枢兴奋作用增强,作用时间延长,从而发挥促进递质释放的作用,如间羟胺和麻黄碱。
若引入其它烷基则活性降低或消失。
5、氨基上有无取代基及取代基的大小对α和β受体的选择有影响。
无取代基的去甲肾上腺素主要为α受体效应,对β受体作用微弱;甲基取代的肾上腺素,α和β受体都有作用;异丙基取代的异丙肾上腺素则主要为β受体效应,α受体作用极弱。
由此可见氨基上随着取代基的增大,α受体效应减弱,β受体效应增强。
拟肾上腺素药的体内代谢主要由两种酶所催化。
一种是儿茶酚氧位甲基转移酶(COMT),催化3位酚羟基的甲基化;一种是单胺氧化酶(MAO),催化氧化脱胺反应。
外源性肾上腺素和体内肾上腺素髓质分泌的肾上腺素,在体内很快被肝脏和其它组织的COMT催化形成间甲肾上腺素,再被MAO催化形成3-甲氧基-4-羟基扁桃酸。





拟肾上腺素药肾上腺素受体激动药与肾上腺素受体结合,激动受体,产生肾上腺素样的作用。
它们都是胺类,而作用又与兴奋交感神经的效应相似,故又称拟交感胺类,其基本化学结构是β-苯乙胺。
α受体激动药α1,α2受体激动药去甲肾上腺素Noradrenaline正肾上腺素【药理作用】非选择性激动α1和α2受体,与肾上腺素比较在某些器官其α作用比肾上腺素略弱,对心脏β1受体作用较弱,对β2受体几无作用。
1.血管激动血管的α1受体,使血管收缩,主要是使小动脉和小静脉收缩。
皮肤粘膜血管收缩最明显,其次是对肾脏血管的收缩作用。
此外脑、肝、肠系膜甚至骨骼肌的血管也都呈收缩反应。
冠状血管舒张,这主要由于心脏兴奋,心肌的代谢产物(如腺苷)增加,从而舒张血管所致,同时因血压升高,提高了冠状血管的灌注压力,故冠脉流量增加。
在一定情况下,也可激动血管壁的去甲肾上腺素能神经突触前α2受体,抑制递质的释放。
2,心脏作用较肾上腺素为弱,激动心脏的β1受体,使心肌收缩性加强,心率加快,传导加速,心搏出量增加。
在整体情况下,心率可由于血压升高而反射性减慢。
过大剂量,心脏自动节律性增加,也会出现心律失常,但较肾上腺素少见。
拟肾上腺素药基本作用的比较3.血压小剂量滴注时由于心脏兴奋,收缩压升高,此时血管收缩作用尚不十分剧烈,故舒张压升高不多而脉压加大。
较大剂量时,因血管强烈收缩使外周阻力明显增高,故收缩压升高的同时舒张压也明显升高,脉压变小。
【剂量与用法】休克:见第九章第六节去甲肾上腺素。
上消化道出血:口服 1mg~3mg(用适量的冰生理盐水稀释)的去甲肾上腺素,可迅速而安全地控制上消化道大出血。
这是由于它的局部作用、使上消化道粘膜血管剧烈收缩而发挥止血作用。
4.其他对机体代谢的影响较弱,只有在大剂量时才出现血糖升高。
对中枢作用也较肾上腺素为弱。
【临床应用】1.休克目前去甲肾上腺素类血管收缩药在休克治疗中已不占主要地位,仅限于某些休克类型如早期经原性休克及药物中毒引起的低血压等,用去甲肾上腺素静脉滴注,使收缩压维持在12kPa左右,以保证心、脑等重要器官的血液供应。